




Autosomal recessive spinocerebellar ataxia type 8 (ARCA1/SCAR8) is caused by mutations of the SYNE1 gene. The disease was initially described in families from Quebec (Canada) with a phenotype of pure cerebellar syndrome, but in recent years has been reported with a more variable clinical phenotype in other countries. Cases have recently been described of muscular dystrophy, arthrogryposis, and cardiomyopathy due to SYNE1 mutations.
ObjectiveTo describe clinical and molecular findings from 4 patients (3 men and one woman) diagnosed with ARCA1/SCAR8 from 3 Spanish families from different regions.
Material and methodsWe describe the clinical, paraclinical, and genetic results from 4 patients diagnosed with ARCA1/SCAR8 at different Spanish neurology departments.
ResultsOnset occurred in the third or fourth decade of life in all patients. After 15 years of progression, 3 patients presented pure cerebellar syndrome, similar to the Canadian patients; the fourth patient, with over 30 years’ progression, presented vertical gaze palsy, pyramidal signs, and moderate cognitive impairment. In all patients, MRI studies showed cerebellar atrophy. The genetic study revealed distinct pathogenic SYNE1 mutations in each family.
ConclusionsARCA1/SCAR8 can be found worldwide and may be caused by many distinct mutations in the SYNE1 gene. The disease may manifest with a complex phenotype of varying severity.
La ARCA1/SCAR8 es un heredoataxia recesiva causada por mutaciones en el gen SYNE1, que fue descrita inicialmente en familias francocanadienses (Quebec) con un síndrome cerebeloso puro. En la actualidad se describe cada vez más este tipo de ataxia en otras partes del mundo y con un fenotipo muy variable. Recientemente se han notificado casos de distrofia muscular, artrogriposis y miocardiopatía por mutaciones de este gen.
ObjetivosDescribir los hallazgos clínicos y moleculares en 3 familias españolas de diferente origen geográfico, las primeras en las que se confirmó el diagnóstico de ARCA1/SCAR8 con análisis molecular.
Material y métodosEvaluación clínica, pruebas paraclínicas y estudio genético en 4 pacientes (3 varones y una mujer), diagnosticados en distintos servicios de neurología españoles.
ResultadosLos síntomas cerebelosos comenzaron en todos los casos en la tercera-cuarta décadas. Tras 15 años de evolución, 3 pacientes presentaban un síndrome cerebeloso puro similar a la descripción original, mientras que un paciente con más de 30 años de evolución presentaba también parálisis de mirada vertical, afectación piramidal y moderado deterioro cognitivo. El estudio de resonancia magnética mostró en todos los casos atrofia restringida al cerebelo. La secuenciación de SYNE1 permitió identificar distintas variantes patogénicas en cada familia.
ConclusionesLa ARCA1/SCAR8 tiene una distribución mundial con una gran diversidad de mutaciones en SYNE1. Además de ataxia puede dar lugar a un complejo fenotipo de inicio y gravedad muy variable.
Autosomal recessive spinocerebellar ataxias (ARCA or SCAR) are a broad, heterogeneous group of neurodegenerative disorders. They can manifest as pure or complex cerebellar syndrome and may be associated with symptoms including intellectual disability, oculomotor abnormalities, pyramidal and extrapyramidal symptoms, and peripheral neuropathy.1–3 Some subtypes present characteristic laboratory findings that facilitate diagnosis.1–3 The most frequent autosomal recessive ataxias include Friedreich ataxia (FA), ataxia telangiectasia, ataxia with vitamin E deficiency, abetalipoproteinaemia, ataxia with oculomotor apraxia (AOA), and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS); all of these are associated with a complex phenotype.1–3
Autosomal recessive cerebellar ataxia type 1 (ARCA1 or SCAR8, OMIM #610743) was first described in families from Beauce and Bas-St-Laurent (Quebec, Canada). The disease is caused by mutations in the spectrin repeat containing nuclear envelope 1 (SYNE1) gene, located on chromosome 6.4,5 The gene includes 146 exons and encodes a giant protein comprising 8797 amino acids, known as nuclear envelope spectrin repeat protein 1 (nesprin 1). The protein is expressed in Purkinje cells, the olivary bodies, and in myocytes; it has 4 domains (one presenting the spectrin-like structure characteristic of membrane-anchored proteins) and plays an important role in maintaining the structure of the cell, as it fixes the nuclear lamina to the cytoskeleton and contributes to the organisation of cytoplasmic organelles.6–8
ARCA1 has also been described in patients from Brazil, Japan, the United Kingdom, Saudi Arabia, and, in a recent multi-centre study using massive sequencing techniques, in families from France, Germany, Belgium, Italy, and Algeria. As a result, it is now considered a globally distributed hereditary ataxia. While ARCA1 was first reported to be associated with a pure cerebellar syndrome, more recent studies have contributed to the knowledge of its phenotype, and it is now known that the disease can present at a wide range of ages as a multisystemic disease with signs of upper and lower motor neuron involvement, musculoskeletal involvement, and cognitive impairment.9
This study presents a comparative description of the phenotype of the disease in the first 3 Spanish families diagnosed with ARCA1.
Patients and methodsFour patients with cerebellar ataxia were attended at the neurology departments of Complexo Hospitalario Universitario de Santiago de Compostela (La Coruña), Hospital Clínico San Carlos (Madrid), and Hospital Universitario Virgen del Rocío (Seville).
In all cases, the following tests were performed before definitive diagnosis of ARCA1: (a) blood analysis (complete blood count; systematic biochemistry; total protein test; lipid profile; vitamins E, B9, and B12; alpha-fetoprotein; ceruloplasmin; copper; lactic, pyruvic, and phytanic acid; cholestanol; chitotriosidase; CCL18 [PARC]; and an antibody panel including ANA, anti-Ro, anti-La, anti-thyroid, anti-gliadin, and anti-GAD antibodies) and urine analysis (elements and sediment, copper); (b) neurophysiological studies (multimodal evoked potentials, motor and sensory nerve conduction velocity); (c) ophthalmological examination; (d) neuroimaging studies with brain and spinal cord MRI, and in one case brain 18F-deoxiglucose positron emission tomography (18FDG-PET); and (e) genetic studies (FA, AOA1, AOA2, SCA1-7, SCA17, NPC1, and NPC2).
Definitive diagnosis was reached by studying the SYNE1 gene using next generation sequencing techniques (ataxia panel) followed by Sanger sequencing to confirm the mutations identified.
ResultsLaboratory analyses (intended to rule out ataxias with biomarkers or susceptible to disease-modifying treatment), ophthalmological examination, neurophysiological studies, and previous genetic studies all yielded normal or negative results.
Table 1 summarises the main clinical data and the pathogenic SYNE1 variants detected (NM_033071.3) in 4 patients from 3 different families. The patients are described below.
Clinical data and SYNE1 mutations.
| Family/patient | Sex | Age of onset/current age | Symptoms | SYNE1 variants detecteda |
|---|---|---|---|---|
| 1/ 1 | M | 27/40 | Predominantly appendicular pure cerebellar syndrome | 1) c.13045C>T; p.R4349* |
| 2) c.25114C>T; p.R8372* | ||||
| 1/ 2 | W | 22/35 | Predominantly axial pure cerebellar syndrome | 1) c.13045C>T; p.R4349* |
| 2) c.25114C>T; p.R8372* | ||||
| 2/ 3 | M | 33/47 | Predominantly axial pure cerebellar syndrome | 1) c.17099_c.17100del; p.I5700Tfs**11 |
| 2) c.4555C>T; p.Q1519* | ||||
| 3/ 4 | W | 35/72 | Cerebellar-plus syndrome (dysphagia, conjugate vertical gaze palsy; pyramidal signs) | 1) c.16177.2A>G |
| 2) c.16177.2A>G |
M: man; W: woman.
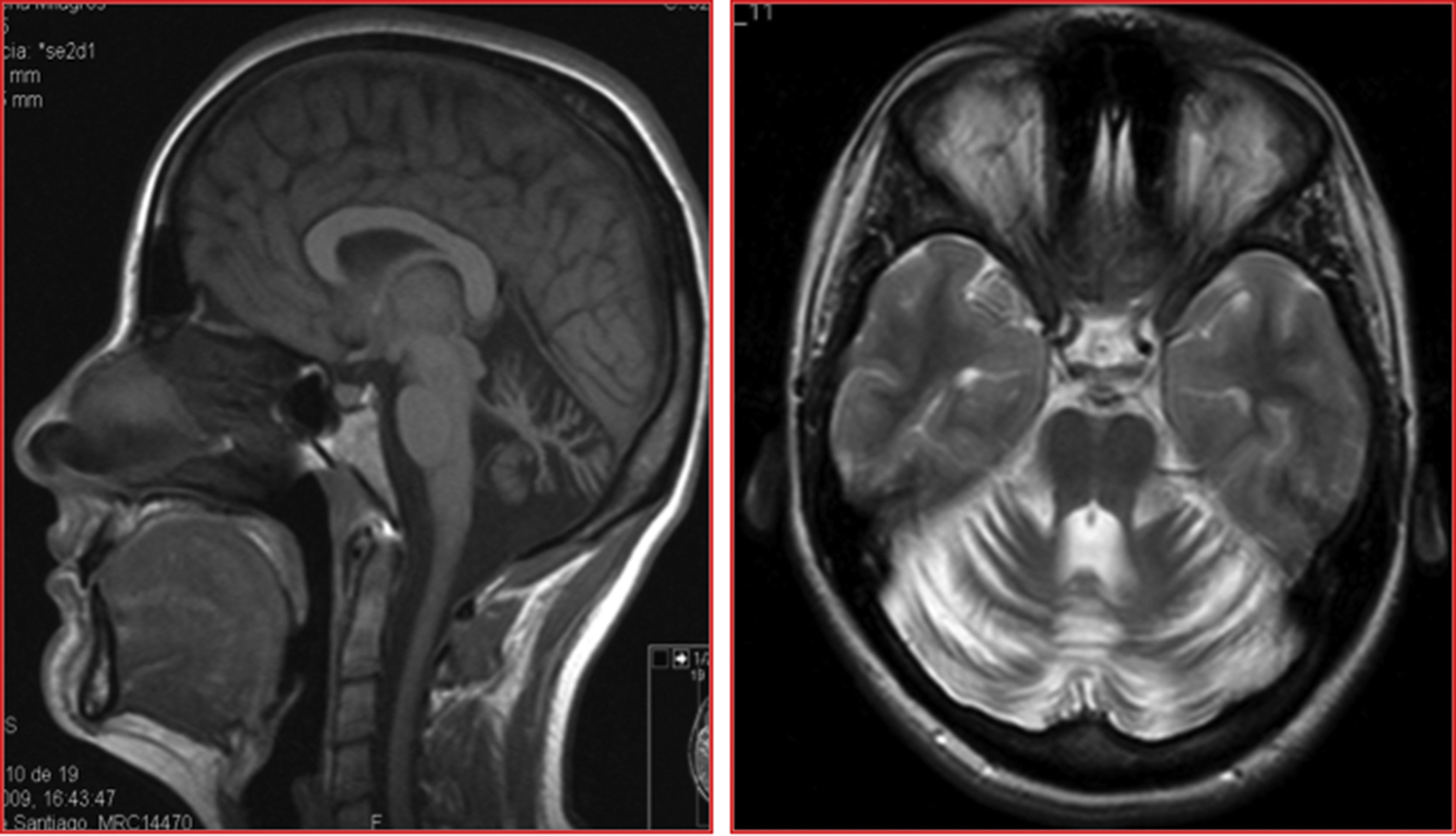
The patients were siblings, a man of 40 years of age and a woman of 35, born to healthy, non-consanguineous parents, in Galicia, Spain. There were no known cases of similar conditions or other related neurological symptoms in the family. In both patients, disease onset occurred in the third decade of life; initial symptoms were instability and dysarthria, followed by slowly progressing loss of limb coordination. After more than 12 years of progression, they were able to walk unsupported. The sister received treatment for moderate anxiety and depression. Both patients presented dysarthria with scanning speech, persistent bidirectional horizontal gaze-evoked nystagmus, appendicular ataxia, and, to a greater extent, truncal ataxia, preventing tandem gait. Tendon reflexes were preserved and plantar reflexes were flexor. In both cases, brain MRI detected marked diffuse atrophy of the cerebellum, with the brainstem and cerebral white matter being unaffected (Fig. 1).

The genetic study identified variants c.13045>T; p.R4349* (exon 77) and c.25114C>T; p.R8372* (exon 140) in both siblings; the first variant was inherited from their father and the second from their mother. Both variants are predicted to cause premature stop codons, resulting in a truncated protein. To date, these variants have not been described in other patients in the literature; their frequency in the gnomAD database is extremely low (0.00005-0.000008).
Family 2: patient 3The patient was a 47-year-old man born to non-consanguineous parents in Andalusia, Spain. Disease onset occurred at the age of 34 years, with impaired speech production; months later, he experienced difficulty descending stairs, and subsequently presented instability while walking and riding a bicycle. At the age of 40 years, he began to lose coordination in the upper limbs. He did not present diplopia, dysphagia, or cognitive impairment. The patient’s brother had developed similar symptoms in the third decade of life, and committed suicide shortly thereafter. A paternal first cousin, a woman of 50 years of age, also presented ataxia, but it was not possible to examine her. Examination of the patient revealed dysarthria with scanning speech, mild ocular movement anomaly in the form of hypermetric saccades, appendicular ataxia, and, to a greater extent, truncal ataxia, which prevented tandem gait. MRI revealed diffuse cerebellar atrophy.
The genetic study revealed 2 truncating SYNE1 variants, which were classed as probably pathogenic: c.17099_c.17100del; p.I5700Tfs*11 (exon 90), a deletion causing a frame shift, and c.4555C>T; p.Q1519* (exon 35), which causes a premature stop codon. Both variants were found in trans. Neither has previously been described either as a mutation or as a polymorphism in the population databases consulted (dbSNP, gnomAD, 1000G).
Family 3: patient 4The patient was a 72-year-old woman whose parents were first cousins, born in Andalusia, Spain. She presented difficulty walking, which had progressed since the age of 35 years. She was the youngest of 6 siblings, and the only one to present ataxia. The initial symptom was gait instability, followed by dysarthria and subsequently loss of limb coordination. More recently, she reported urinary incontinence and mild memory complaints. Examination revealed mixed dysarthria (spastic and cerebellar), supranuclear vertical gaze palsy, bidirectional horizontal nystagmus, hypometric horizontal saccades, dysmetria and dysdiadochokinesia in all 4 limbs, moderate spasticity, and tendon hyperreflexia in the lower limbs, with ankle clonus and bilateral Babinski sign. At the time of examination, she was unable to stand or walk, and used a wheelchair. The neuropsychological examination showed deficits in learning and verbal memory consolidation and moderate impairment of visuospatial function; she did not meet diagnostic criteria for dementia.
The genetic study revealed that the patient was homozygous for a previously described pathogenic SYNE1 variant (c.16177-2A>G), located in intron 84, affecting a splice acceptor site; the variant is predicted to cause the loss of exon 85, disrupting the normal maturation of the mRNA. We also detected a missense variant of uncertain significance: c.9736C>G; p.Q3246E, registered as rs149901087 on the dbSNP151 database, with a population frequency of 0.02%-0.06%. Three of 7 bioinformatic analysis systems predicted that the variant may be deleterious. As we would expect, all the patient’s daughters were heterozygous carriers of both variants.
Structural neuroimaging revealed diffuse atrophy of the cerebellum. We also performed a brain 18FDG-PET study, which showed diffuse cerebellar hypometabolism with no metabolic alterations in other brain regions.
DiscussionTo our knowledge, these are the first 4 cases of ARCA1/SCAR8 in Spanish patients. In all cases, the disease initially presented with truncal ataxia and/or dysarthria in the third or fourth decade of life. Three patients, with disease progression times of around 15 years, presented slowly progressive pure cerebellar syndromes, with pancerebellar atrophy on MRI studies and no evidence of polyneuropathy in neurophysiological studies. The fourth patient, a woman with a gait disorder of over 35 years’ progression, presented pyramidal tract involvement, supranuclear oculomotor palsy, and moderate cognitive impairment, in addition to the cerebellar syndrome. One interesting finding was that the brain 18FDG-PET study performed in this patient revealed hypometabolism in the cerebellum only. In other words, in the first 2 decades of progression, our patients’ clinical symptoms overlap with those described in patients from Quebec,4,5 where this form of ataxia was first described, and where it constitutes the third leading cause of autosomal recessive ataxia, after ARSACS and FA.10
In 2013, a Japanese study reported the first cases not originating in Canada.11 One patient presented onset during childhood, with motor neuron disease associated with ataxia, and 2 presented similar symptoms to the French-Canadian patients. It should be noted that in Japan, SCA36 (a dominant hereditary ataxia caused by an intronic expansion in NOP56) presents with more extensive motor neuron involvement than that observed in the “Costa da Morte” SCA36 ataxia described in Spain.12,13 In the same year, 2 cases of ARCA1 were diagnosed in patients from Brazil and France, who presented mutations not described in the families from Quebec.14
A study conducted in the United Kingdom included 196 cases of sporadic or autosomal recessive ataxia and identified SYNE1 mutations in 4 patients from 3 families (from Turkey, Sri Lanka, and the United Kingdom).15 Three patients presented pure cerebellar syndrome, whereas the patient of Turkish origin presented marked pyramidal tract involvement. The authors analyse and describe all the mutations reported at the time of their study, and postulate that greater proximity of the mutations to the 3′ end of the gene is associated with greater predisposition to motor neuron involvement.15 However, we did not observe motor neuron signs in our patients from family 1, who presented a mutation in exon 140. In 2016, an extensive multi-centre study (including centres from various European countries and Algeria) was published, which included 434 index patients with the most prevalent forms of SCA and FA.9 Exome sequencing detected 23 patients (5%) from unrelated families, in whom they identified 35 pathogenic variants (34 not previously described). The clinical phenotypes reported in that study consisted of pure cerebellar syndrome in 19% of cases and complex forms in the remaining patients. The authors observed motor neuron disease in 58% of patients, intellectual development disorder in 10%, and respiratory dysfunction secondary to brainstem involvement in 3 cases. The mean age of onset was 22 years (range, 6-40), lower than that observed in our patients and in Canadian studies. The authors conclude that ARCA1 should be considered a frequent cause of autosomal recessive ataxia.9 We should underscore the fact that none of the Spanish patients described in this study carried any of the mutations reported in previous publications.
More recently, the spectrum of manifestations associated with SYNE1 mutations has continued to expand. Given the intense cerebellar atrophy observed, the cerebellar hypoplasia and intellectual disability in some of these patients are particularly interesting, as they suggest a phenotypic spectrum ranging from neonatal manifestations to adult onset.16 In a family from Saudi Arabia with ARCA1, MRI showed lesions suggestive of multiple sclerosis, but without oligoclonal bands in the cerebrospinal fluid and with normal visual evoked potentials.17 A new family has been described in Japan that does not present associated motor neuron disease.18 Heterozygous SYNE1 mutations have been reported in several cases resembling Emery-Dreifuss muscular dystrophy, arthrogryposis, and dilated cardiomyopathy.19,20
In conclusion, ARCA1 seems to be emerging as one of the most prevalent forms of autosomal recessive ataxia. SYNE1 mutations are one of the most frequent causes of pure cerebellar syndrome with onset in young adults, whether sporadic or with suspected autosomal recessive inheritance. Suspicion should be even stronger in the event of neuroimaging findings of marked pancerebellar atrophy and absence of polyneuropathy or biochemical markers of other types of ARCA. However, given the clinical overlap between ARCA1 and many other entities, we consider massive sequencing (ataxia panel or whole-exome sequencing) to be the most suitable diagnostic approach in the majority of patients.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Arias M, Mir P, Fernández-Matarrubia M, Arpa J, García-Ramos R, Blanco-Arias P, et al. Heredoataxia cerebelosa recesiva ARCA1/SCAR8: primeras familias detectadas en España. Neurología. 2022;37:257–262.
