




La ARCA1/SCAR8 es una heredoataxia recesiva causada por mutaciones en el gen SYNE1, que fue descrita inicialmente en familias francocanadienses (Quebec) con un síndrome cerebeloso puro. En la actualidad se reporta cada vez más este tipo de ataxia en otras partes del mundo y con un fenotipo muy variable. Recientemente se han notificado casos de distrofia muscular, artrogriposis y miocardiopatía por mutaciones de este gen.
ObjetivosDescribir los hallazgos clínicos y moleculares en 3 familias españolas de diferente origen geográfico, las primeras en las que se confirmó el diagnóstico de ARCA1/SCAR8 con análisis molecular.
Material y métodosEvaluación clínica, pruebas paraclínicas y estudio genético en 4 pacientes (3 varones y una mujer), diagnosticados en distintos servicios de neurología españoles.
ResultadosLos síntomas cerebelosos comenzaron en todos los casos en la tercera-cuarta décadas. Tras 15 años de evolución, 3 pacientes presentaban un síndrome cerebeloso puro similar a la descripción original, mientras que un paciente con más de 30 años de evolución presentaba también parálisis de mirada vertical, afectación piramidal y moderado deterioro cognitivo. El estudio de resonancia magnética mostró en todos los casos atrofia restringida al cerebelo. La secuenciación de SYNE1 permitió identificar distintas variantes patogénicas en cada familia.
ConclusionesLa ARCA1/SCAR8 tiene una distribución mundial con una gran diversidad de mutaciones en SYNE1. Además de ataxia puede dar lugar a un complejo fenotipo de inicio y gravedad muy variable.
Autosomal recessive spinocerebellar ataxia type 8 (ARCA1/SCAR8) is caused by mutations of the SYNE1 gene. The disease was initially described in families from Quebec (Canada) with a phenotype of pure cerebellar syndrome, but in recent years has been reported with a more variable clinical phenotype in other countries. Cases have recently been described of muscular dystrophy, arthrogryposis, and cardiomyopathy due to SYNE1 mutations.
ObjectiveTo describe clinical and molecular findings from 4 patients (3 men and one woman) diagnosed with ARCA1/SCAR8 from 3 Spanish families from different regions.
Material and methodsWe describe the clinical, paraclinical, and genetic results from 4 patients diagnosed with ARCA1/SCAR8 at different Spanish neurology departments.
ResultsOnset occurred in the third or fourth decade of live in all patients. After 15 years of progression, 3 patients presented pure cerebellar syndrome, similar to the Canadian patients; the fourth patient, with over 30 years’ progression, presented vertical gaze palsy, pyramidal signs, and moderate cognitive impairment. In all patients, MRI studies showed cerebellar atrophy. The genetic study revealed distinct pathogenic SYNE1 mutations in each family.
ConclusionsARCA1/SCAR8 can be found worldwide and may be caused by many distinct mutations in the SYNE1 gene. The disease may manifest with a complex phenotype of varying severity.
Las ataxias espinocerebelosas autosómicas recesivas (ARCA o SCAR) constituyen un amplio y heterogéneo grupo de trastornos neurodegenerativos. Su fenotipo puede consistir en un síndrome cerebeloso puro o complejo, asociado a discapacidad intelectual, alteraciones oculomotoras, clínica derivada de afectación piramidal, extrapiramidal y neuropatía periférica, entre otros1–3. En algunos subtipos se pueden detectar alteraciones características en pruebas de laboratorio que facilitan el diagnóstico1–3. La ataxia de Friedreich (AF), la ataxia telangiectasia (AT), la ataxia por déficit de vitamina E (AVED), la abetalipoproteinemia, las ataxias con apraxia oculomotora (AOA) y la ataxia espástica de Charlevoix-Saguenay (ARSACS) son las ataxias recesivas más frecuentes; todas ellas cursan con un fenotipo complejo1–3.
La ataxia cerebelosa de herencia recesiva tipo 1 (ARCA1 o SCAR8, MIM# 610743) fue descrita inicialmente en familias de Beauce y Bas-St. Laurent (Quebec-Canadá). Está causada por mutaciones en el gen spectrin repeat containing nuclear envelope 1 (SYNE1), localizado en el cromosoma 64,5. Este gen se compone de 146 exones que codifican una proteína gigante de 8.797 aminoácidos denominada nesprina 1 (nuclear envelope spectrin repeat protein 1); esta proteína se expresa en células de Purkinje, de la oliva bulbar y también en miocitos, posee 4 dominios —uno de ellos con estructura de tipo espectrina característica de proteínas de anclaje a membranas— y tiene una importante función de mantenimiento de la estructura celular ya que fija la lámina nuclear al citoesqueleto y contribuye a la organización de las organelas citoplasmáticas6-8.
La ARCA1 también ha sido descrita en pacientes de Brasil, Japón, Reino Unido, Arabia Saudí y, en un reciente estudio multicéntrico con técnicas de secuenciación masiva, en familias de Francia, Alemania, Bélgica, Italia y Argelia, por lo que ha pasado a ser considerada una ataxia hereditaria de distribución mundial. Por otra parte, mientras en la descripción inicial se asoció la ARCA1 a un síndrome cerebeloso puro, los trabajos más recientes han permitido expandir el conocimiento de su fenotipo, que puede presentarse en un amplio abanico de edad como enfermedad multisistémica que incluye signos de motoneurona superior e inferior, afectación músculo-esquelética y deterioro cognitivo9.
El objetivo de este trabajo es presentar una descripción comparada de las 3 primeras familias españolas diagnosticadas de ARCA1.
Pacientes y métodosCuatro pacientes con ataxia cerebelosa fueron consultados en los Servicios de Neurología del Complexo Hospitalario Universitario, Santiago de Compostela, La Coruña, del Hospital Clínico de San Carlos, Madrid y del Hospital Universitario Virgen del Rocío, Sevilla.
En todos ellos, antes de llegarse al diagnóstico definitivo de ARCA1, se habían realizado: a) análisis de sangre (hematimetría, bioquímica sistemática, proteinograma, lipidograma, vitaminas E y B12, ácido fólico, alfa-fetoproteína, ceruloplasmina, cobre, ácidos láctico, pirúvico y fitánico, colestanol, quitotriosidasa, CCL18/PARC, panel de anticuerpos incluyendo ANA, anti-Ro, anti-La, anti-tiroideos, anti-gliadina y anti-GAD) y orina (elemental y sedimento, cobre); b) estudios neurofisiológicos (potenciales evocados multimodales, velocidades de conducción nerviosa motora y sensitiva; c) examen oftalmológico; d) estudios de neuroimagen con resonancia magnética (RM) cerebral y medular; en un caso tomografía de emisión de positrones con 18F-desoxiglucosa (18FDG-PET) cerebral, y e) estudios genéticos (AF, AOA1, AOA2, SCA1-7, SCA17, NPC1 y NPC2).
El diagnóstico definitivo se alcanzó estudiando el gen SYNE1 mediante técnicas de secuenciación de nueva generación (panel de ataxias) y confirmación de las mutaciones identificadas mediante secuenciación convencional tipo Sanger.
ResultadosLos análisis (encaminados a descartar ataxias con marcadores bioquímicos y susceptibles de tratamiento modificador del curso evolutivo), el examen oftalmológico, los estudios neurofisiológicos y los estudios genéticos realizados anteriormente resultaron normales o negativos.
En la tabla 1 se resumen los datos clínicos principales y las variantes patogénicas detectadas en el gen SYNE1 (NM_033071.3) en 4 pacientes pertenecientes a 3 familias distintas, que describimos a continuación, por separado:
Datos clínicos y mutaciones detectadas en el gen SYNE1
| Familia: paciente | Sexo | Edad inicio/edad actual | Cuadro clínico | Variantes encontradas en SYNE1a |
|---|---|---|---|---|
| I: 1 | V | 27/40 | Síndrome cerebeloso puro de predominio apendicular | 1) c.13045C>T; p.R4349*2) c.25114C>T; p.R8372* |
| I: 2 | M | 22/35 | Síndrome cerebeloso puro de predominio axial | 1) c.13045C>T; p.R4349*2) c.25114C>T; p.R8372* |
| II: 3 | V | 33/47 | Síndrome cerebeloso puro de predominio axial | 1) c.17099_c.17100del; p.I5700Tfs**112) c.4555C>T; p.Q1519* |
| III: 4 | M | 35/72 | Síndrome cerebeloso plus: disfagia, paresia de mirada conjugada vertical, piramidalismo | 1) c.16177.2A>G2) c.16177.2A>G |
M: mujer; V: varón.
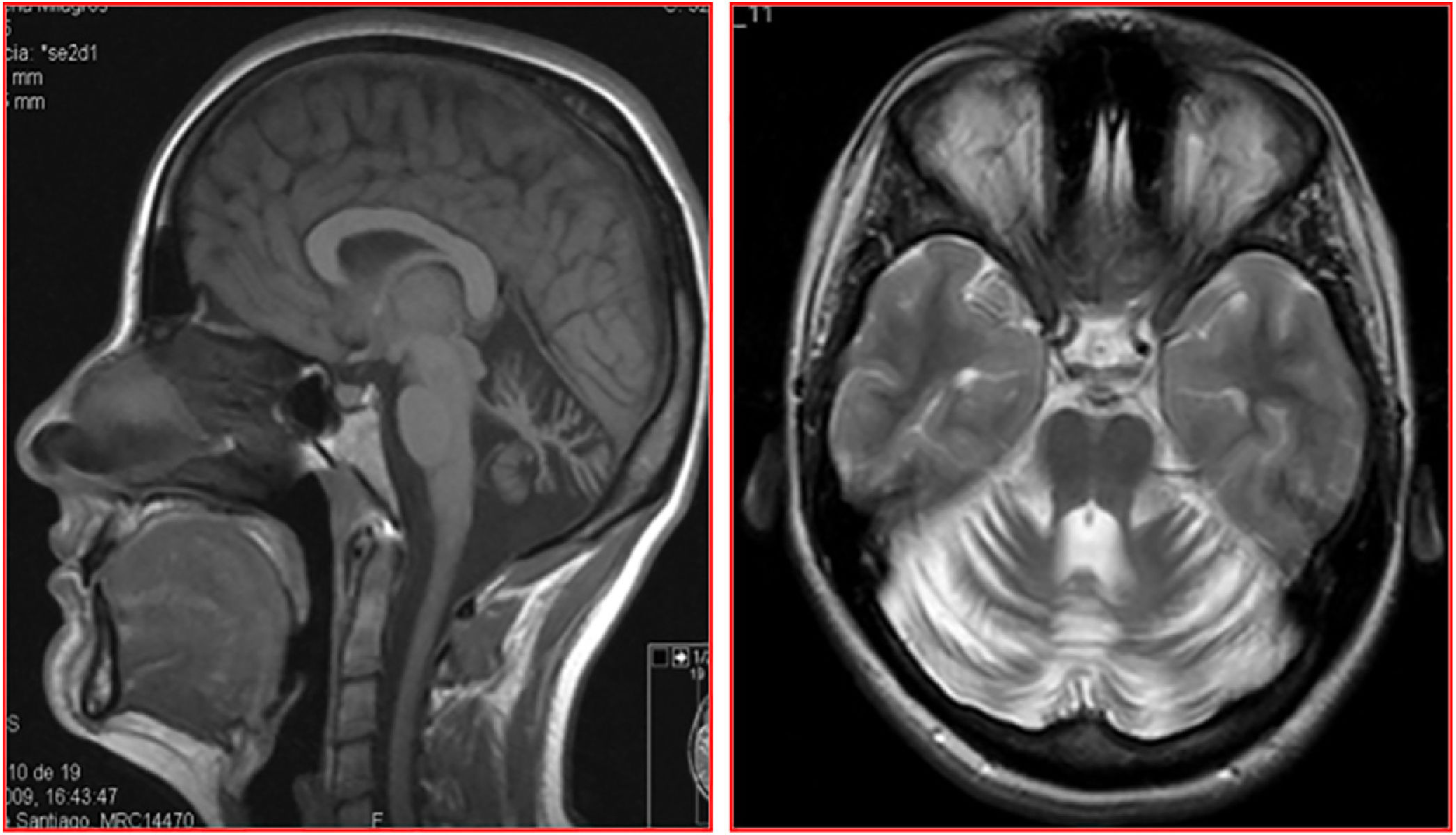
Constituida por 2 hermanos, un varón de 40 años y una mujer de 35, de padres sanos y sin consanguinidad, nacidos en Galicia, España; no hay conocimiento de casos similares ni otros síntomas neurológicos relacionados en la familia. En ambos, la enfermedad se inició en la tercera década de la vida, con desequilibrio y disartria, seguidos de torpeza y descoordinación en extremidades de progresión lenta. Después de una evolución superior a 12 años caminaban sin necesidad de apoyo. La hermana recibió tratamiento por síndrome ansioso-depresivo moderado. Ambos presentaban disartria con palabra escandida, nistagmo inagotable evocado en la mirada horizontal bidireccional, ataxia apendicular y, en mayor grado, de línea media, con imposibilidad para realizar la marcha en tándem. Los reflejos tendinosos estaban preservados con reflejos cutáneo-plantares flexores. En el estudio de RM cerebral se detectó, en ambos casos, una marcada atrofia difusa de cerebelo, sin atrofia del tronco cerebral ni alteraciones de la sustancia blanca encefálica (fig. 1).
 y T2 (axial) del paciente I: 1: se observa atrofia difusa del cerebelo con morfología normal del tronco cerebral y sin lesiones de sustancia blanca.")
El estudio genético permitió identificar en ambos hermanos las variantes c.13045>T;p.R4349* (exón 77) y c.25114C>T;p.R8372* (exón 140), la primera heredada del padre y la segunda de la madre. Ambas variantes previsiblemente producen un codón de parada prematuro y generan una proteína truncada. Hasta la fecha no se han descrito estas variantes en otros pacientes en la literatura; solo se encuentran depositadas en la base de datos gnomAD con frecuencias extremadamente bajas (0.00005-0.000008).
Familia II: paciente 3Varón de 47 años, hijo de padres no consanguíneos y nacidos en Andalucía, España. Comenzó a los 34 años con problemas para articular el lenguaje, meses después apreció dificultad para bajar escaleras, y posteriormente desequilibrio al caminar y montar en bicicleta. A los 40 años comenzó a mostrar torpeza de extremidades superiores. No presenta diplopía, disfagia ni deterioro cognitivo. Un hermano del paciente, que inició en la tercera década un cuadro similar, se suicidó poco tiempo después. Una prima hermana por línea paterna, de 50 años, presentaba también ataxia, pero no ha podido ser explorada. En la exploración se observó disartria con palabra escandida, leve alteración de movimientos oculares en forma de sacadas hipermétricas, ataxia apendicular y, en mayor grado, de línea media, con imposibilidad para realizar la marcha en tándem. En el estudio de RM se detectó atrofia cerebelosa difusa.
En el estudio genético se identificaron en el gen SYNE1 dos variantes truncantes, probablemente patogénicas: c.17099_c.17100del; p.I5700Tfs*11 (exón 90), deleción que altera el patrón de lectura, y c.4555C>T; p.Q1519* (exón 35), que origina un codón de parada. Ambas variantes están en trans. Ninguna ha sido descrita previamente ni como mutaciones ni como polimorfismos en bases de datos poblacionales consultadas (dbSNP, gnomAD, 1000G).
Familia III: paciente 4Se trata de una mujer de 72 años, hija de padres primos hermanos y nacidos en Andalucía, España. Presentaba dificultad progresiva para caminar desde los 35 años. Es la menor de 6 hermanos y solo ella padece ataxia. Sus síntomas iniciales fueron desequilibrio al caminar, seguido de disartria y, posteriormente, torpeza de extremidades. Más recientemente refería incontinencia urinaria y quejas leves de falta de memoria. En la exploración se objetivó disartria mixta (espástica y cerebelosa), parálisis de mirada vertical supranuclear, nistagmo en la mirada horizontal bidireccional, sacadas horizontales hipométricas, dismetría y disdiadococinesia en las 4 extremidades, espasticidad moderada e hiperreflexia tendinosa en miembros inferiores con clonus aquíleo y signo de Babinski bilateral. En el momento de ser evaluada no era capaz de mantener bipedestación ni caminar, y precisaba silla de ruedas. En el estudio neuropsicológico mostraba rendimiento deficitario en el aprendizaje y consolidación de la memoria verbal y moderada afectación de la función visuoespacial, sin criterios de demencia.
El estudio genético detectó en homocigosis en el gen SYNE1 una variante patogénica previamente descrita (c.16177-2A>G), localizada en el intrón 84, que afecta a un sitio aceptor de splicing, que previsiblemente ocasionará la pérdida del exón 85 y perturbará la maduración normal del mRNA. Se detectó asimismo una variante missense de significado incierto c.9736C>G; p.Q3246E, registrada en la base de datos dbSNP151 como rs149901087, con una frecuencia poblacional entre el 0.02-0.06%. Tres de 7 sistemas de análisis bioinformático predicen que podría tratarse de una variante deletérea. Todas las hijas de la paciente resultaron, como cabía esperar, portadoras en heterocigosis de ambas variantes.
En la neuroimagen estructural se pudo constatar la existencia de atrofia cerebelosa difusa. A la paciente se le realizó además un estudio de 18FDG-PET cerebral, que mostró la existencia de hipometabolismo cerebeloso difuso, sin afectación metabólica de otras regiones encefálicas.
DiscusiónEn el presente trabajo se describen los 4 primeros casos, en nuestro conocimiento, de pacientes españoles con ARCA1 (SCAR8). Todos los pacientes iniciaron la clínica con ataxia de línea media y/o disartria en la tercera-cuarta década de vida. En 3 de ellos, con una evolución cercana a los 15 años, la enfermedad cursa con un síndrome cerebeloso puro y lentamente progresivo, con atrofia pancerebelosa en el estudio de RM, y sin evidencia de polineuropatía en estudios neurofisiológicos. En el cuarto paciente, una mujer con trastorno de la marcha de más de 35 años de evolución, además del síndrome cerebeloso se constató afectación piramidal y parálisis oculomotora supranuclear y deterioro cognitivo moderado. Es interesante subrayar que el estudio de 18FDG-PET cerebral de esta paciente mostró hipometabolismo restringido únicamente al cerebelo. Es decir, nuestros pacientes presentan en las 2 primeras décadas de evolución un cuadro clínico superponible al descrito en los pacientes francocanadienses de Quebec4,5, donde esta variedad de ataxia fue inicialmente descrita y es la tercera causa de ataxia recesiva tras ARSACS y AF en dicha región10.
En 2013 se publicaron en Japón los primeros casos de ARCA1 no originarios de Canadá11. Uno de ellos de inicio infantil y con enfermedad de motoneurona asociada a la ataxia, y 2 pacientes similares a los francocanadienses. A destacar que en Japón, la propia SCA36 (heredoataxia dominante producida por expansión intrónica en el gen NOP56) presenta un cuadro clínico con una afectación más extensa de motoneuronas que la forma de SCA36-Ataxia da Costa da Morte, descrita en España12,13. También en ese mismo año se diagnosticaron 2 casos de ARCA1 en sendos pacientes originarios de Brasil y Francia, que presentaban cada mutaciones no descritas en las familias de Beauce-Quebec-Canadá14.
En un estudio de 196 casos de ataxia esporádica o recesiva realizado en el Reino Unido y dirigido a buscar mutaciones de SYNE1, se encontraron 4 pacientes de 3 familias (Turquía, Sri Lanka y Reino Unido)15. El fenotipo consistía en un síndrome cerebeloso puro en 3 de los pacientes, mientras que se acompañaba de marcado piramidalismo en el caso de origen turco. Los autores analizan y detallan todas las mutaciones publicadas hasta la realización de su trabajo y postulan que una mayor cercanía de las mutaciones al extremo final 3’ del gen conllevaría mayor predisposición a afectación de motoneuronas15. Sin embargo, nosotros no hemos observado signos de motoneurona en la familia I del presente estudio, con una mutación en el exón 140 de la proteína. También en 2016 se publicó un extenso estudio multicéntrico (diversos países europeos y Argelia), que incluyó 434 pacientes-índice con cuadros ataxia en los que se habían excluido las SCA más prevalentes y la AF9. Mediante secuenciación total del exoma se detectaron 23 pacientes (5%) de otras tantas familias no relacionadas, en los que se identificaron 35 variantes patogénicas (34 no descritas previamente). Los fenotipos clínicos reportados en dicho estudio consistieron en un síndrome cerebeloso puro en el 19% de los casos y complicado en el resto. Los autores observaron enfermedad de motoneurona en el 58%, trastorno del desarrollo intelectual en el 10% y disfunción respiratoria por afectación de tronco cerebral en 3 casos. La edad media de comienzo de los síntomas fue de 22 años (rango de 6-40), inferior a la de nuestros casos y a los canadienses. Los investigadores concluyen que la ARCA1 debe pasar a ser considerada una causa frecuente de ataxia recesiva9. A reseñar que ninguno de los pacientes españoles que describimos en el presente trabajo era portador de mutaciones descritas en las publicaciones previas.
Más recientemente, el espectro de manifestaciones asociadas a SYNE1 no ha dejado de crecer. Dada la intensa atrofia cerebelosa observada en estos pacientes, es de especial interés la descripción de pacientes con hipoplasia cerebelosa y discapacidad intelectual, que sugieren una continuidad fenotípica desde las manifestaciones neonatales hasta la edad adulta16. En una familia procedente de Arabia Saudí diagnosticada de ARCA1 el estudio de RM mostró lesiones sugestivas de esclerosis múltiple, pero sin bandas oligoclonales en el LCR y con normalidad de potenciales evocados visuales17. En Japón, se ha notificado una nueva familia pero, en este caso, sin enfermedad de neurona motora asociada18. Por otra parte, se han notificado mutaciones de SYNE1 en heterocigosis en varios casos similares a la distrofia muscular de Emery-Dreifuss, en pacientes con artrogriposis congénita y en otros con miocardiopatía dilatada19,20.
En conclusión, la ARCA1 parece que se está posicionando como una de las formas más frecuentes de ataxia recesiva. Ante un cuadro cerebeloso puro de inicio en un adulto joven, esporádico o sugestivo de herencia recesiva, las mutaciones en SYNE1 se encuentran entre las causas más frecuentes. Aumentan el grado de sospecha el hallazgo de una atrofia pancerebelosa marcada en la neuroimagen, así como ausencia de polineuropatía y marcadores bioquímicos de otras ARCA. Sin embargo, la variabilidad clínica constatada en ARCA1 conlleva un solapamiento con otras muchas entidades, por lo que en nuestra opinión el abordaje diagnóstico mediante secuenciación masiva —panel de genes causantes de ataxia o WES— es más adecuado en la mayoría de los pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses relacionado con el contenido del presente trabajo.

