




Las enfermedades mitocondriales son de herencia materna, y dentro de las más prevalentes se encuentra el síndrome de MELAS. Las características clínicas de la enfermedad son miopatía, encefalopatía, convulsiones, acidosis láctica y episodios similares a accidentes cerebrovasculares (ACV). Su presentación típica es en adultos jóvenes, adolescentes y niños, siendo mucho más raro su inicio en adultos mayores.
Casos clínicosSe presentan los casos clínicos de 2 pacientes femeninas, hija y madre, de 31 y 75 años, respectivamente. Ambas son de baja estatura y presentaban hipoacusia como síntoma compartido previamente. La primera con inicio en la adultez temprana, caracterizado por varios episodios que asemejaban un ACV, acidosis láctica y convulsiones; y la segunda con encefalopatía, y un episodio similar a ACV. Ambas con RMN cerebral que mostraba lesiones extensas sin respetar territorios vasculares específicos y una evolución con mejoría imagenológica posterior. La más joven realizó test genético que confirmó la enfermedad mitocondrial. Su madre comenzó después, por lo que la sospecha diagnóstica fue dirigida a MELAS. Para valorar las secuelas, se realizaron test neuropsicológicos, ambos con evidencia de trastorno neurocognitivo menor (hija) y mayor (madre).
ConclusiónEl síndrome MELAS debe sospecharse en pacientes con episodios recurrentes que asemejen un ACV e imágenes que se resuelven posteriormente. Recalcamos la necedad de una sospecha diagnostica elevada, aun en presentaciones tardías luego de la sexta década de la vida. Pese a no existir una terapéutica específica, se puede controlar la enfermedad con suplementación de aminoácidos y pautas nutricionales estrictas para evitar crisis metabólicas que exacerben el cuadro. Debe considerarse que no existe tratamiento probado para detener la progresión de la enfermedad.
Mitochondrial diseases are maternally inherited, and the most prevalent is MELAS syndrome. The clinical features of the disease are myopathy, encephalopathy, seizures, lactic acidosis, and stroke-like episodes (SLEs). Its typical presentation is in young adults, adolescents and children, its debut in older adults being much rarer.
Clinical casesWe present clinical cases of 2 female patients, daughter and mother, aged 31 and 75, respectively. Both are short of stature and had hearing loss as a previously shared symptom. The first with debut in early adulthood characterized by several episodes that resembled a stroke, lactic acidosis and seizures; and the second with encephalopathy, and a SLE. Both with brain MRI that showed extensive lesions without respecting specific vascular territories and an evolution with subsequent imaging improvement. The youngest performed a genetic test that confirmed mitochondrial disease. His mother debuted later, so the diagnostic suspicion was directed to MELAS. To assess the consequences, neuropsychological tests were performed, both with evidence of showing minor (daughter) and major (mother) neurocognitive disorder.
ConclusionMELAS syndrome should be suspected in patients with recurrent SLEs and images that subsequently resolve. We emphasize the foolishness of a high diagnostic suspicion, even in late presentations after the 6th decade of life. Although there is no specific therapy, the disease can be controlled with amino acid supplementation and strict nutritional guidelines to avoid metabolic crises that exacerbate the condition. It should be considered that there is no proven treatment to stop the progression of the disease.
Las enfermedades mitocondriales son un grupo heterogéneo de afecciones que se caracterizan fisiopatológicamente por la disfunción de la cadena respiratoria mitocondrial, clave para la producción de ATP, siendo el MELAS su forma más frecuente. El síndrome fue descrito por primera vez en el año 1984 por Pavlakis et al.1 y su acrónimo en inglés (Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-like episodes) se compone de 3 características que definen la enfermedad: encefalopatía mitocondrial, acidosis láctica y eventos «stroke-like». Es transmitida exclusivamente por vía materna2 y es el resultado de anomalías ya sea del ADN nuclear o del mitocondrial, siendo esta última la mutación más frecuente3.
Es una enfermedad rara, con una incidencia de 1,6 por cada 10.000 personas, con una presentación que ocurre típicamente durante la infancia, por lo que la mayoría de los casos son diagnosticados antes de los 20 años de edad3,4. Sin embargo, pueden existir casos de presentación tardía diagnosticados luego de los 40 años de edad5.
Las mitocondrias son organelos intracelulares con un número intracelular proporcional a los requerimientos energéticos de cada célula; por tal motivo, las neuronas y las células musculares, las cuales tienen una alta necesidad energética, comparten la característica de poseer un alto número de mitocondrias en su composición celular. Las mitocondrias son el pilar de la producción energética a través de la producción de ATP por medio de fosforilación oxidativa6,7.
Molecularmente la patogénesis se explica por la sustitución de adenina por guanina en el nucleótido 32433 del gen MT-TL1. Esta mutación se encuentra en el 80% de los pacientes, existiendo otras variantes menos frecuentes. Una de las características de esta mutación es su carácter heteroplásmico, distribuyéndose de manera azarosa entre el ADN mitocondrial y generando la diversidad fenotípica de la enfermedad.
Las mutaciones han sido localizadas en el gen ATN t Leu del ADN mitocondrial –tNRALeu–, siendo identificadas más de 16 mutaciones posibles causantes de la enfermedad. Esta mutación genera una traducción mitocondrial alterada que desencadena una disminución de la síntesis de proteínas mitocondriales y la consecuente caída de la producción de energía mitocondrial. La deficiencia de ATP produce la falla multiorgánica y una proliferación mitocondrial. Esta última se genera a nivel del músculo liso y las células endoteliales de los vasos sanguíneos, produciendo una angiopatía con alteración de la microvasculatura que contribuye a las complicaciones de la enfermedad, especialmente los episodios «stroke-like»8.
Se plantea que también existe una deficiencia de óxido nítrico multifactorial, lo que podría contribuir a las complicaciones de la enfermedad. La deficiencia de óxido nítrico a nivel del endotelio vascular resulta en una alteración de la perfusión sanguínea en la microvasculatura, contribuyendo a los síntomas de la enfermedad, además de producir un agravamiento de la acidosis láctica9.
Las manifestaciones clínicas de la enfermedad son muy heterogéneas; esto se debe a 3 factores principales: la coexistencia entre el ADN mutado y normal (heteroplasmia), la distribución variable del ADN mitocondrial mutado en los tejidos, y la respuesta variable que posee cada tejido al estrés oxidativo.
Estas manifestaciones variables hacen que en algunas ocasiones el diagnóstico sea un desafío. Presentamos 2 casos de MELAS en madre e hija que experimentaron fenotípicamente episodios «stroke-like».
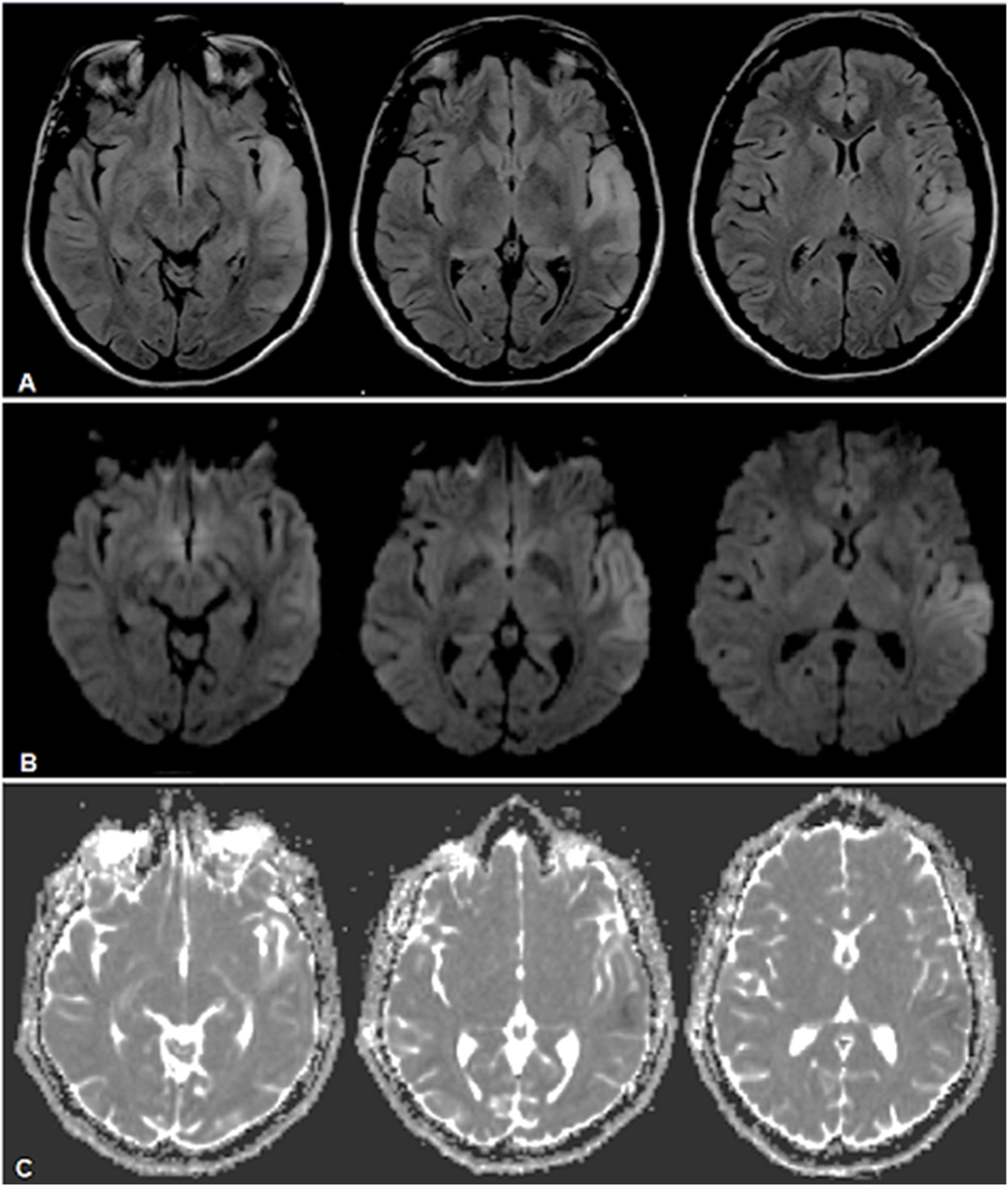
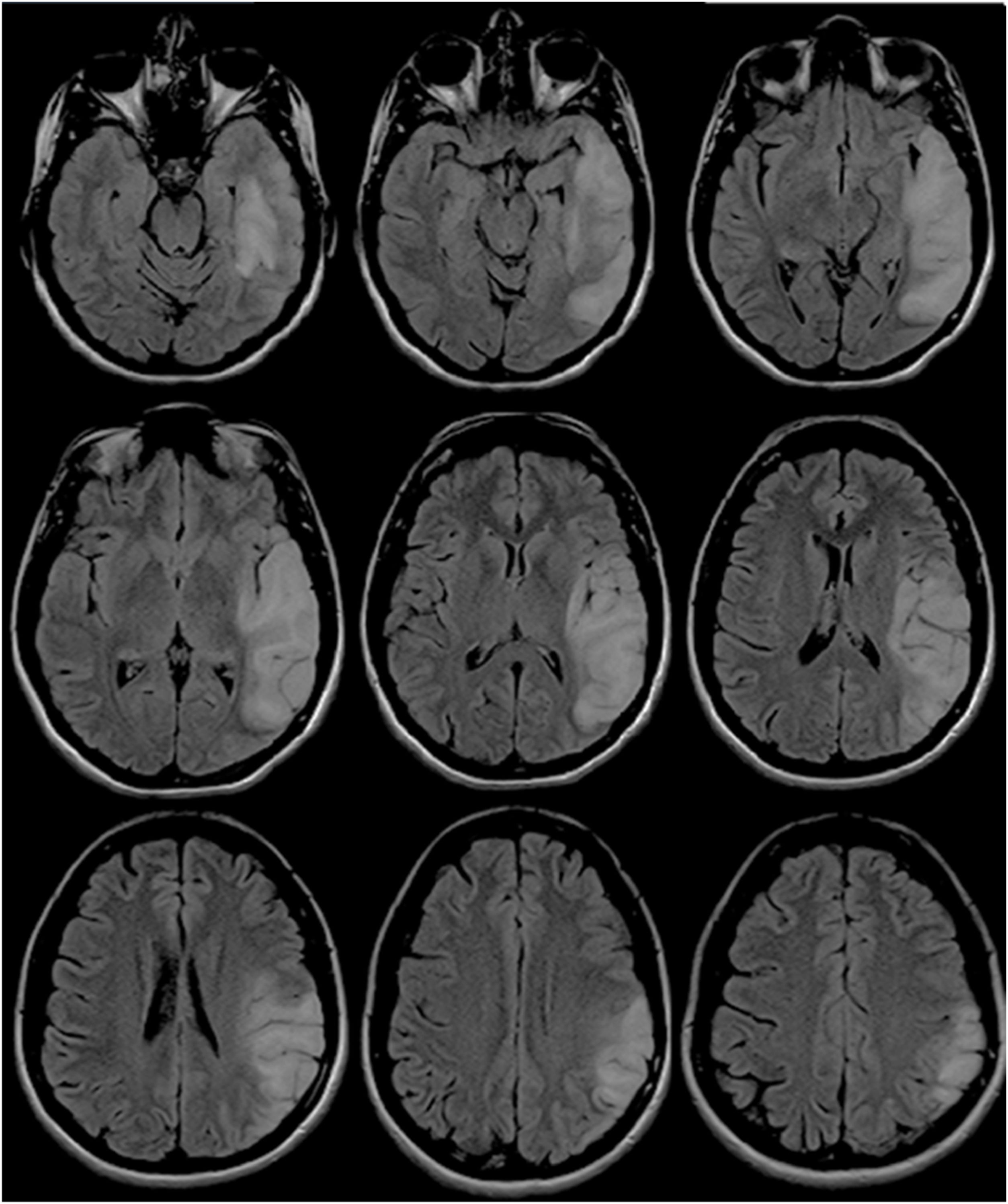
Caso clínico 1Paciente femenina de 31 años con antecedentes de hipoacusia neurosensorial profunda bilateral desde los 27 años con requerimiento de audífonos, y aborto retenido a los 26 años. Consultó inicialmente por afasia de expresión. Se realizó una resonancia magnética nuclear (RMN) cerebral que evidenció una lesión hiperintensa temporal izquierda (fig. 1). Se interpretó en primera instancia como un accidente cerebrovascular isquémico (ACVi) de etiología en estudio, destacándose causas de hipercoagulabilidad, reumatológicas, cardiológicas y neoplásicas. Posteriormente, intercurrió con parestesias periorales y del hemicuerpo derecho, con nueva RMN cerebral con espectroscopia que demostró extensión de la lesión previa (fig. 2), marcado pico de lactato y leve inversión de la relación colina/NAA. El estudio metabólico arrojó una creatina fosfocinasa de 156UI/L, lactacidemia de 2,2 y ácido pirúvico de 4,6 pre y postingesta de 60mg de glucosa, lactacidemia de 3,72mmol/L, aldolasa de 5,8mmol/L, aminoácidos plasmáticos elevados a expensas de fenilalanina, taurina, leucina, isoleucina, metionina, valina, treonina, prolina, glicina, ornitina y glutamina; ácidos orgánicos en orina con presencia de 3-hidroxibutírico y acetoacético (cetosis), y creatinina en orina de 227,4mg/dL. Por último, ácido láctico en líquido cefalorraquídeo (LCR) de 3,59mg/dL.
, con restricción parcial a nivel cortical a predominio posterior indicativo de áreas de edema citotóxico y otras áreas de efecto T2 a predominio yuxtacortical temporal anterior, indicativo de edema vasogénico en secuencias DWI (B) y ADC (C).")
RMN de cerebro donde se observa lesión hiperintensa a nivel temporal lateral izquierdo en secuencias FLAIR (A), con restricción parcial a nivel cortical a predominio posterior indicativo de áreas de edema citotóxico y otras áreas de efecto T2 a predominio yuxtacortical temporal anterior, indicativo de edema vasogénico en secuencias DWI (B) y ADC (C).

Se clasificó el evento como episodios «stroke-like» en paciente con síndrome de MELAS por diagnóstico clínico y molecular, con confirmación genética posterior que reveló una variante patogénica heteroplásmica del 35% m3243A>G (tNRALeu).
Ulteriormente se realizó una infusión programada de arginina intravenosa. En dicha infusión la paciente intercurrió con crisis convulsivas focales con generalización secundaria y posteriormente subintrantes, que mejoraron en el transcurso de la hospitalización y tratamiento acorde.
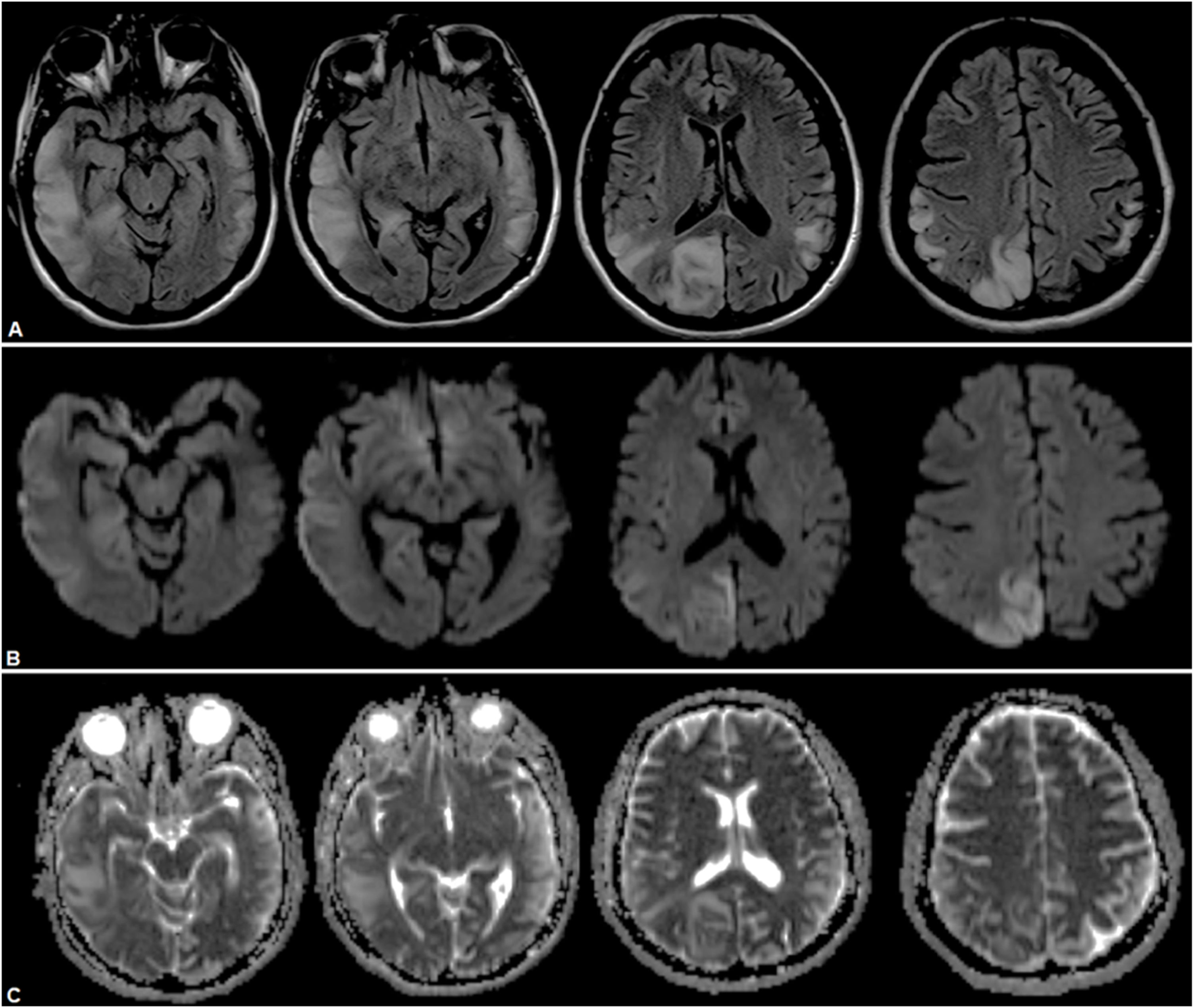
Dos años después del diagnóstico presentó 3 recaídas en 3 meses consecutivos. La primera estaba caracterizada por tinnitus, cefalea y alucinaciones; la segunda, por hemianopsia izquierda; y la tercera, por desorientación en espacio y ataxia crural izquierda. Se realizó una nueva RMN, que expuso la extensión lesional a predominio cortical comprometiendo ahora los lóbulos frontal, parietal, temporal y occipital derechos (fig. 3). Recibió en cada ocasión carga y mantenimiento con arginina intravenosa durante 5 días.
 a nivel temporooccipitoparietal derecha con restricción a la secuencia DWI asociado a edema vasogénico (B y C).")
No volvió a presentar recaídas, continuó en tratamiento anticomicial con levetiracetam, aminoácidos por vía oral, coenzima Q10 y seguimiento estricto por nutrición y equipo de enfermedades metabólicas. Como secuelas presenta deterioro neurocognitivo menor multidominio, no amnésico, e hipoacusia que corrige con el uso de audífonos.
Caso clínico 2Paciente femenina de 75 años de edad, de baja estatura, con antecedentes de queja cognitiva de un año de evolución, hipoacusia bilateral desde los 68 años, pérdida de peso y disminución de la ingesta los últimos 6 meses, cuya hija presenta síndrome de MELAS (caso 1). Se presentó con cuadro clínico de 72h de evolución caracterizado por pérdida de la habilidad para realizar actividades cotidianas y trastorno de la marcha, episodios aislados de conversaciones incoherentes y leve inestabilidad postural e incontinencia urinaria. En el examen físico presentó episodio febril asociado a hemianopsia homónima izquierda y ataxia braquiocrural izquierda.
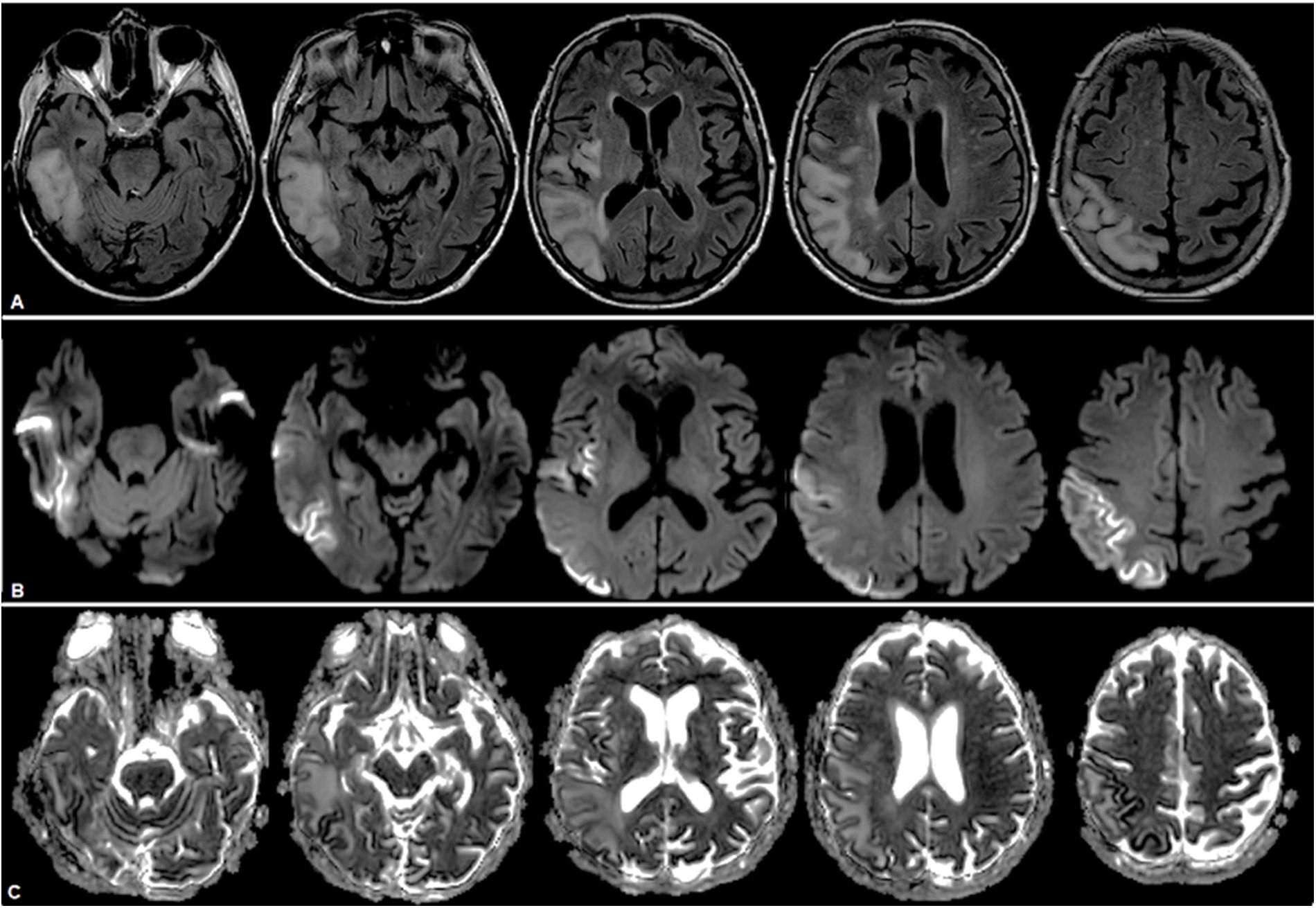
Se confirmó infección por COVID-19, por lo que se mantuvo en aislamiento. Se realizó estudio cerebral por RMN, que mostró una lesión córtico-subcortical temporoparietooccipitoinsular derecha, con focos de restricción cortical y edema vasogénico subcortical, y lesión cerebelosa basal izquierda (fig. 4). No se logró la realización de una espectroscopia por poca tolerancia de la paciente al estudio. Por sospecha de ACVi se estudió la etiología, descartando causas cardíacas y afectación de grandes vasos. Por antecedente familiar de enfermedad mitocondrial se solicitó estudio metabólico, con lactacidemia de 2,21mmol/L, dosaje de acilcarnitinas normales, aminoácidos en sangre sin alteraciones, carnitina total elevada de 92,8nmol/L (35-84), carnitina libre sin alteraciones, ácidos grasos no esterificados de 0,59mmol/L, betahidroxibutirato 0,62mmol/L (0,10-0,30). Se realizó el diagnóstico de episodio «stroke-like» en paciente con síndrome de MELAS por diagnóstico clínico. No se realizó test genético, ya que se tomó en cuenta el antecedente de su descendencia de primer grado.
, con restricción a predominio cortical indicativo de áreas de edema citotóxico y áreas de efecto T2 a predominio yuxtacortical, indicativo de edema vasogénico en secuencias DWI (B) y ADC (C).")
RMN de cerebro donde se observa lesión hiperintensa a nivel temporoparietooccipitoinsular derecha en secuencias FLAIR (A), con restricción a predominio cortical indicativo de áreas de edema citotóxico y áreas de efecto T2 a predominio yuxtacortical, indicativo de edema vasogénico en secuencias DWI (B) y ADC (C).
Se administró arginina intravenosa durante 5 días, con evolución favorable. Se realizó test neuropsicológico, que mostró trastorno neurocognitivo mayor de grado leve con síntomas conductuales asociados.
Continúa con suplementación de aminoácidos y coenzima Q10 por vía oral, seguimiento estricto por nutrición y equipo de enfermedades metabólicas. No volvió a presentar recaídas por el momento.
DiscusiónEl ACVi es infrecuente en personas jóvenes, por lo que ante estos eventos es necesario considerar enfermedades menos frecuentes pero relevantes en este rango etario. Existen estudios que evidencian que en pacientes menores de 45 años con ACVi en el circuito posterior, un 6% tendrá una mutación del ADN mitocondrial en la posición 32439,10.
Dentro de las 6 manifestaciones cardinales del síndrome se encuentran: «stroke-like», crisis epilépticas, acidosis láctica, fibras rojas rasgadas, intolerancia al ejercicio y comienzo de los síntomas antes de los 40 años. A estas manifestaciones se adiciona la baja estatura y el retraso del crecimiento en las poblaciones juveniles, y la pérdida auditiva y la diabetes mellitus en las formas en la adultez11. Estas manifestaciones se correlacionan con los tejidos con mayores requerimientos energéticos y, por lo tanto, con mayor número de mitocondrias, como el músculo esquelético y cardíaco, el riñón, el páncreas, el hígado, la retina y el sistema nervioso central.
La miopatía deviene de un defecto grave en la cadena respiratoria de los mioblastos y se manifiesta por debilidad muscular, intolerancia al ejercicio y, en algunos casos, atrofia muscular. El síntoma más característico es la debilidad, que suele ser proximal y progresiva. No suele ser el motivo de consulta del paciente, ya que este síntoma suele ser atribuido a otras causas.
La encefalopatía típica del síndrome MELAS suele ser progresiva y se caracteriza por la presencia de crisis convulsivas. Estas crisis recurrentes contribuyen al daño cortical con atrofia y los consecuentes trastornos cognitivos a largo plazo.
La alteración metabólica con el posterior fallo energético producido en la enfermedad afecta la oxidación mitocondrial con la consiguiente producción de ácido láctico con el resultado de acidosis metabólica con GAP aumentado. En condiciones basales esta situación suele ser subclínica, pero en condiciones en las que aumenta la demanda energética corporal, como en infecciones o ayuno, se puede desencadenar una descompensación hasta acidosis metabólica12.
Los casos presentados tuvieron como manifestación cardinal episodios «stroke-like», el síntoma más frecuente de la enfermedad y que permite su diferenciación de otras enfermedades mitocondriales multisistémicas como el MERF y el síndrome de Kearns-Sayre. Ocurren por una disfunción de la fosforilación oxidativa a nivel del parénquima cerebral y un aumento de la producción de radicales libres con la resultante vasoconstricción que causarían los cuadros «stroke-like». Se caracterizan por episodios habitualmente reversibles de focalidad neurológica, como trastorno del lenguaje, trastornos visuales, paresia, cefalea, crisis convulsivas o alteraciones del estado mental, como somnolencia, obnubilación o coma.
Las banderas de alarma para el correcto diagnóstico de un evento similar al ACVi incluyen episodios que se acompañan frecuentemente de crisis epilépticas, las lesiones no corresponden a un territorio vascular específico, son más frecuentes en el territorio posterior, se expanden hacia regiones contiguas y tanto los hallazgos en neuroimágenes como los síntomas suelen ser reversibles.
Frente a alguna de estas características, en el contexto de un paciente con epidemiología compatible (mujer, joven), los diagnósticos diferenciales de episodios «stroke-like» deben incluir el síndrome MELAS, puesto que constituye un reto diagnóstico complejo ya que puede presentarse a una edad variable y sus síntomas pueden afectar a casi cualquier órgano. Sumatoriamente a su heteroplasmia, hacen del MELAS una enfermedad de difícil sospecha diagnóstica.
Yatsuga et al. formularon una serie de criterios diagnósticos en el año 2012. Estos se dividen en 2 categorías: la A, con 5 ítems (cefaleas/vómitos, crisis epilépticas/encefalopatía, hemiplejía, ceguera cortical/hemianopsia o lesión focal aguda observada en TAC o RMN), y la categoría B, con 3 ítems (niveles elevados de ácido láctico en suero y/o LCR/deficiencia de actividad de enzimas mitocondriales, anomalías mitocondriales en la biopsia muscular y mutaciones genéticas definidas relacionadas con MELAS). Establecieron como criterios definitivos la presencia de al menos 2 elementos de la categoría A y 2 elementos de la categoría B, y como criterios indicativos de MELAS la presencia de al menos un elemento de la categoría A y 2 elementos de la categoría B13.
Nuestra paciente más joven reunía criterios para MELAS definitivo, ya que presentó 2/2 características de la categoría A y 2/2 características de la categoría B en el momento del diagnóstico (focalidad neurológica y alteración por RMN, acidosis láctica y test genético con mutación específica).
La paciente mayor reunía criterios para MELAS sugestivo, presentando 2/2 características de la categoría A y 1/2 características de la categoría B (encefalopatía, hemianopsia, alteración por RMN y test genético extrapolado de la hija) en el momento de su inicio.
En ninguna se realizó biopsia muscular, ya que se contaba con el resultado de la mutación genética mitocondrial.
Dentro del estudio químico en pacientes con sospecha de MELAS, deben buscarse dirigidamente niveles elevados de lactato en sangre, así como el cociente lactato/piruvato. La normalidad del lactato en sangre no descarta el diagnóstico de esta enfermedad, ya que este puede estar elevado solo en LCR. La creatina cinasa puede estar elevada, mientras que la coenzima Q10 puede encontrarse disminuida. La paciente mayor del segundo caso clínico no contaba con lactacidemia elevada, y no se completó el estudio en el LCR por la negativa de la paciente.
Otro de los estudios complementarios que pueden aportar al diagnóstico es la biopsia muscular, donde característicamente se observan fibras rojo-rasgadas en la tinción tricrómico de Gomori7. La presencia de estas fibras se explica por la proliferación mitocondrial. La biopsia es positiva en el 85% de los casos, pero no excluye la enfermedad14.
En cuanto a los hallazgos por RMN cerebral, son orientativos a síndrome de MELAS la predilección por regiones posteriores, incluyendo la corteza de los lóbulos parietales, temporales y occipitales, con restricción de la difusión cortical giriforme; pueden ser hipointensas o hiperintensas en ADC. En T2 y FLAIR suelen estar edematosas con pérdida de diferenciación córtico-subcortical. La espectroscopia suele mostrar un pico de lactato y una disminución de N-acetil aspartato15,16.
El análisis genético constituye la prueba diagnóstica de certeza. El estudio molecular debe realizarse en los tejidos con mayor afectación, ya que se ha demostrado, comparando muestras de sangre y músculo, que el porcentaje de mutación es mucho más bajo en sangre, hasta incluso niveles indetectables. En cambio, en el músculo la mutación es claramente positiva17.
El origen tan tardío de la enfermedad en la segunda paciente podría explicarse por ser portadora de una baja carga de mutaciones. Recordemos que la heteroplasmia es una característica de la enfermedad, lo que la hace complejamente heterogénea y de curso evolutivo muy diverso. En la literatura existen pocos reportes de casos mayores de 50 años, entre ellos el de un paciente japonés de 74 años con una mutación en el gen 14453G mtDNA18 y una mujer de 58 años con la mutación 3243A>G mtDNA19, en quienes en un principio se pensó en una encefalitis autoinmune y se logró llegar al diagnóstico con el curso evolutivo de la enfermedad. Hay otros reportes que incluyen la mutación clásica20, y por primera vez Xu et al. describen el síndrome de MELAS con mutación m.14487T>C mtDNA en un paciente de 58 años21. Por lo tanto, inferimos que nuestra paciente es uno de los casos de mayor edad reportados en la literatura científica.
Las opciones terapéuticas actuales para MELAS se basan en la suplementación empírica de multivitamínicos, coenzima Q10, antioxidantes como la vitamina E o el ácido lipídico, la L-carnitina y L-arginina (aminoácido no esencial involucrado en la síntesis de óxido nítrico y la relajación vascular dependiente del endotelio, lo que puede explicar su beneficio, particularmente en la fase aguda de la enfermedad)22. La citrulina y la taurina también han sido utilizadas23. No obstante, los ensayos para una terapia específica continúan y en este momento no hay un tratamiento dirigido que pueda detener la progresión de la enfermedad o evitar recaídas.
ConclusionesDebe sospecharse un síndrome de MELAS en pacientes con episodios recurrentes similares a un ACVi e imágenes cerebrales con lesiones que se resuelven posteriormente, habiendo descartado previamente otros diagnósticos diferenciales. Este diagnóstico debe sospecharse incluso en pacientes con edad tardía de presentación, como demuestra el segundo caso presentado.
La prueba diagnóstica de certeza incluye el estudio genético molecular, y pese a que puede no estar disponible en algunos centros, se puede alcanzar al diagnóstico con la suma de otros criterios.
Incluso cuando no existe una terapéutica específica a la fecha, se puede controlar la enfermedad con suplementación de aminoácidos, multivitamínicos, arginina y pautas nutricionales estrictas para evitar crisis metabólicas que exacerban el cuadro. No hay un tratamiento probado para detener la progresión de la enfermedad y nuevas terapias génicas parecen ser prometedoras para el futuro.
FinanciaciónNinguna.
Conflicto de interesesNinguno para declarar.
