




El síndrome de Cogan es una enfermedad autoinmune rara caracterizada por manifestaciones oculares, auditivas y raramente compromiso sistémico.
Caso clínicoPaciente femenina de 62 años estudiada por eventos cerebrovasculares isquémicos (ACV) sin causa aparente, con aparición posterior de cefalea, pérdida de peso, anacusia bilateral, vértigo y nuevo ACV. Se diagnosticó vasculitis sistémica. Se interpretó el cuadro como Síndrome de Cogan y se inició tratamiento inmunosupresor, con mala respuesta, ya que la paciente falleció posteriormente.
ConclusiónEl ACV como manifestación inicial del síndrome de Cogan es extremadamente raro, por lo que debería ser sospechado ante eventos isquémicos recurrentes sin otra causa aparente.
Cogan's syndrome is a rare autoimmune disease characterized by ocular and auditory manifestations. The key symptoms are interstitial keratitis and sudden hearing loss. Many patients also present systemic compromise, vasculitis being a rare manifestation of the disease.
Case report62-year-old female patient who consulted for transient brachial dysesthesias and after a complete evaluation the diagnosis of ischemic cerebrovascular accident (CVA) of undetermined cause was made. Later, she presented a new cerebrovascular event, for which she was studied again, without pathological findings. In the following two months, the presence of systemic symptoms (headache, weight loss), bilateral hearing loss, episodes of vertigo and a new stroke were added to the clinical picture. Due to suspicion of systemic vasculitis, positron emission tomography was performed with evidence of aortic and femoral vasculitis. The condition was interpreted as Cogan's syndrome and immunosuppressive treatment was started with daily oral meprednisone and monthly cyclophosphamide. Despite treatment, the patient evolved with a new extensive stroke associated with thrombosis of both carotid arteries. Plasmapheresis was performed, with no response since the patient died 4 months after the clinical picture began.
ConclusionThe presentation of systemic manifestations of Cogan's syndrome prior to visual and auditory involvement represent a rare onset of the disease. Likewise, stroke as an initial manifestation is extremely rare, so it should be suspected in cases of repetitive events in patients with no other apparent causes.
El síndrome de Cogan fue descrito por primera vez en 1945 por David Cogan definiéndolo como la asociación de queratitis intersticial y pérdida súbita de la audición1. A partir de un trabajo publicado en 1980 por Haynes se introducen otras formas de presentación posibles y se establecen criterios diagnósticos no universales y no aprobados que, no obstante, ayudan en la práctica diaria al reconocimiento de esta entidad extremadamente poco frecuente2.
Fisiopatológicamente se cree que se trata de una enfermedad autoinmune mediada por autoanticuerpos contra el denominado «péptido Cogan» presente en el suero de algunos pacientes. Este péptido se encontraría en el oído interno y en el endotelio de los vasos sanguíneos3. De esta teoría se desprende que las manifestaciones clínicas responden a afectación ocular, del oído interno y sistémicas. Suele presentarse con pérdida súbita de la audición, asociada a queratitis intersticial. En 70% de los casos se suman síntomas sistémicos como cefalea, artralgias y fiebre4, y en 15-20% pueden encontrarse manifestaciones secundarias a vasculitis5. El accidente cerebrovascular (ACV) no es una manifestación totalmente aceptada como parte de esta patología por la escasa evidencia disponible6. Presentamos un caso clínico de una paciente con ACV como primera manifestación de síndrome de Cogan, sin respuesta al tratamiento y evolución fulminante.
Caso clínicoMujer de 62 años que comienza con disestesias braquiales derechas transitorias. Se realizó laboratorio que evidenció anemia (hemoglobina 10,5 mg/dL) y trombocitosis de 516.000/mm3 y resonancia magnética (RM) de cerebro que mostró lesión isquémica frontal izquierda (fig. 1A). Se interpretó el cuadro como ACV por lo que se completaron estudios con angiotomografía de cerebro y cuello y ecocardiograma transtorácico, con resultado normal, y se inició tratamiento preventivo con antiagregantes y estatinas. Luego de un mes presenta parestesias braquiales derechas transitorias, por lo que se realiza nueva RM de cerebro con hallazgo de lesión isquémica en giro prefrontal izquierdo (fig. 1B). Se realizó ecocardiograma transesofágico con pasaje de burbujas, hemocultivos, serologías para virus de la inmunodeficiencia humana (VIH) y prueba de laboratorio de investigación de enfermedades venéreas (VDRL), Holter de 48 horas y estudio de la coagulación (factor V de Leiden, mutación de protrombina, inhibidor del activador del plasminógeno (PAI), antitrombina III, factor V, VII, VIII, proteína C, resistencia a proteína C, fibrinógeno, anticoagulante lúpico y anticuerpos anticardiolipinas) sin hallazgos de relevancia. Se interpretó el cuadro como ACV isquémico de causa indeterminada.
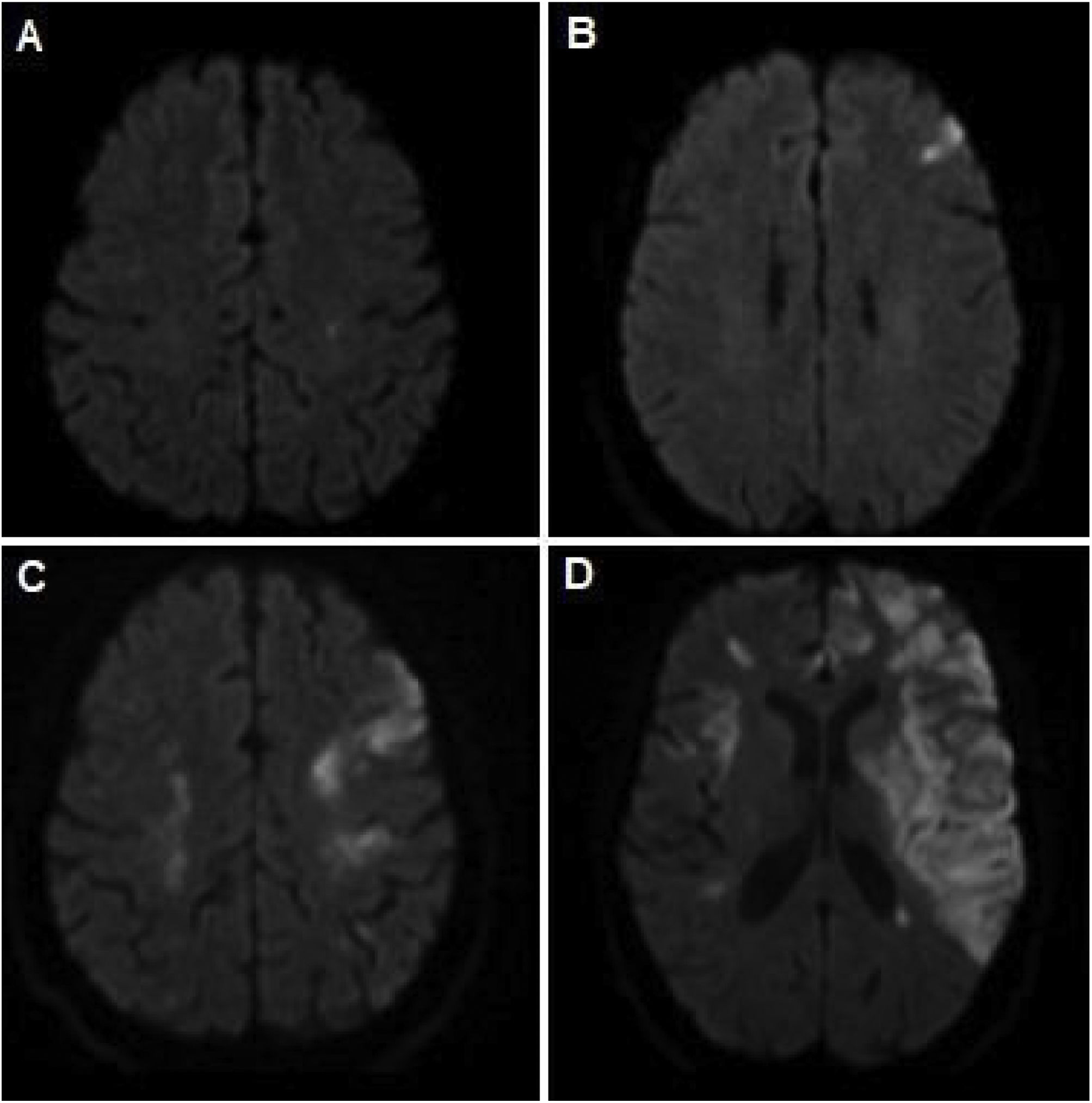
 que evidencia los diferentes eventos isquémicos presentados a lo largo de evolución de la enfermedad. A) Lesión puntiforme a nivel frontal izquierdo. B) Lesión a nivel prefrontal izquierdo. C) Lesiones isquémicas corticales en territorio de ambas arterias cerebrales medias. D) Extensas lesiones isquémicas a nivel del territorio carotídeo izquierdo y derecho.")
RM de cerebro (secuencia de difusión cerebral-DWI) que evidencia los diferentes eventos isquémicos presentados a lo largo de evolución de la enfermedad. A) Lesión puntiforme a nivel frontal izquierdo. B) Lesión a nivel prefrontal izquierdo. C) Lesiones isquémicas corticales en territorio de ambas arterias cerebrales medias. D) Extensas lesiones isquémicas a nivel del territorio carotídeo izquierdo y derecho.
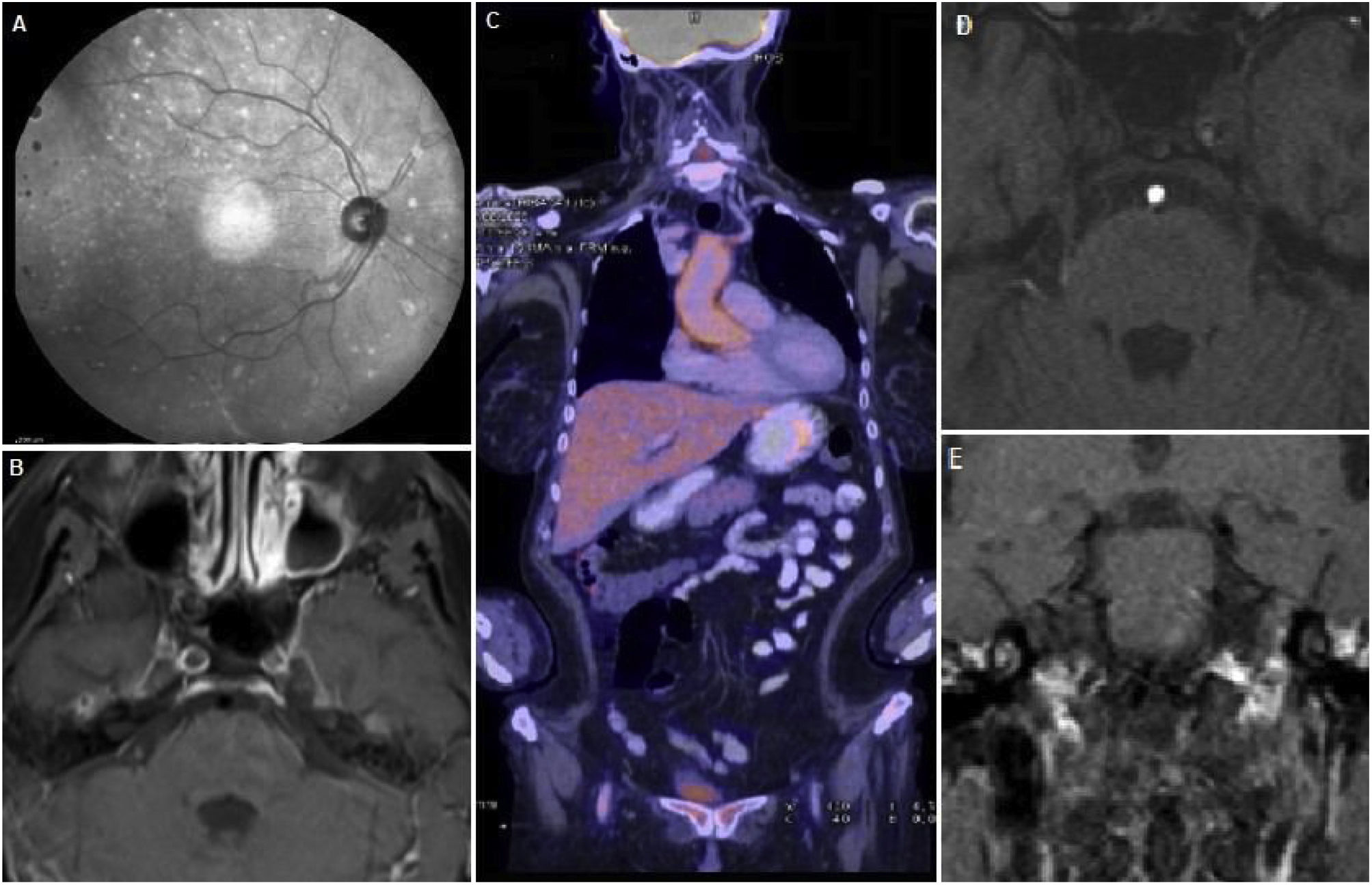
En el lapso de los siguientes dos meses agregó, de forma progresiva, cefalea, pérdida de peso, hipoacusia neurosensorial bilateral, episodios recurrentes de vértigo con RM de conducto auditivo internos que evidenció laberintitis bilateral, esclero-uveítis bilateral (fig. 2A) y nuevas lesiones isquémicas subagudas en territorio limítrofe izquierdo (entre arterias cerebral media y cerebral anterior) manifestadas por trastorno del lenguaje recurrente y transitorio. Se decide su re-internación para ampliar estudios: laboratorio control con progresión de anemia y trombocitosis (6,7 g/dL, punción pulmonar [PL] de 837.000/mm3, respectivamente), elevación de los reactantes de fase aguda (proteína C reactiva [PCR] 271 mg/L, eritrosedimentación [ERS] 81 mm, ferritina sérica 1.228 ng/mL). Se realizó tomografía de tórax, abdomen y pelvis, ecografía transvaginal, marcadores tumorales (antígeno ca 15,3, 19,9 y carcinoembrionario), doppler de arterias temporales y perfil reumatológico (crioglobulinas, proteinograma e inmunoelectroforesis y anticuerpos anticitoplasma de neutrófilo, ADN, nucleares, La, Ro, Smith, ribonucleoproteína, beta 2 glicoproteína, proteína 3 y mieloperoxidasa) normales. Por sospecha de vasculitis sistémica se realizó tomografía con emisión de positrones (PET) que evidenció captación de fluorodesoxiglucosa [FDG] a nivel de aorta ascendente, cayado y torácica y arteria femoral derecha compatibles con vasculitis de grandes vasos (fig. 2C). Se interpretó el cuadro como Síndrome de Cogan probable, por lo que se inició tratamiento con meprednisona 40 mg/día vía oral y ciclofosfamida mensual en pulsos endovenosos. Luego de un mes evolucionó con progresión de la hipoacusia y nuevo evento isquémico, sintomático para paresia braquial derecha y RM cerebro control con nuevas lesiones isquémicas en forma bilateral (fig. 1C) y angiotomografía de cuello con estenosis carotídea moderada bilateral. Se iniciaron cinco pulsos de metilprednisolona 1 g/día. Evolucionó con mutismo y diplejia braquial con RM de encéfalo que con extensa lesión isquémica en ambos territorios carotídeos (fig. 1D). Dada la respuesta tórpida se decide realizar nuevo pulso de metilprednisolona y plasmaféresis, sin respuesta favorable, por lo que fallece en dicha internación a los cuatro meses del inicio del cuadro clínico.
 Fluoroscopia con focos de coroiditis. B) RM cerebro con contraste que evidencia captación de la pared de ambas carótidas internas asociado a ausencia de flujo de las mismas. C) PET con evidencia de captación de FDG a nivel aórtico y femoral derecho. D) AngioRM de cerebro donde se observa ausencia de relleno de ambas carótidas internas. E) RM de conductos auditivos internos con contraste EV, se menciona captación de contraste de ambos laberintos (a predominio izquierdo), compatible con laberintitis.")
Hallazgos sistémicos. A) Fluoroscopia con focos de coroiditis. B) RM cerebro con contraste que evidencia captación de la pared de ambas carótidas internas asociado a ausencia de flujo de las mismas. C) PET con evidencia de captación de FDG a nivel aórtico y femoral derecho. D) AngioRM de cerebro donde se observa ausencia de relleno de ambas carótidas internas. E) RM de conductos auditivos internos con contraste EV, se menciona captación de contraste de ambos laberintos (a predominio izquierdo), compatible con laberintitis.
El síndrome de Cogan, entidad poco frecuente, se caracteriza por la inflamación ocular y síntomas audiovestibulares, que puede asociarse a manifestaciones sistémicas. Presenta una incidencia muy baja que ronda los 300 casos reportados5. Sin predominio de género, afecta principalmente a adultos jóvenes (30 años)5,7 aunque hay casos descritos en los extremos de la vida5.
Clínicamente se caracteriza por la afección ocular y audiovestibular, manifestándose ambas en un lapso de dos años6. El compromiso oftalmológico suele ser el síntoma inicial de la enfermedad y típicamente presenta queratitis intersticial bilateral, aunque puede mostrar otras manifestaciones. El compromiso audiovestibular suele ser posterior al ocular, aunque en algunos casos es la primera manifestación5,8. Lo característico es un síndrome simil Ménière aunque más grave, prolongado y bilateral, pudiendo manifestarse en algunos pacientes con pérdida de audición sin síntomas vertiginosos5. A nivel sistémico puede presentar vasculitis, principalmente de medianos y grandes vasos5,8,7. Está descrito el compromiso cardiovascular, neuromuscular, gastrointestinal y renal. Es común la presencia de otros síntomas generales como fiebre, pérdida de peso, astenia, mialgias y artralgias5. El compromiso sistémico como primera manifestación de la enfermedad, como en el caso de nuestra paciente, es poco frecuente, reportado en 7% de los individuos8. La afección neurológica está descrita en 29% de los pacientes y raramente aparecen previo al compromiso típico ocular y otológico6. Algunas publicaciones sugieren manifestaciones neurológicas asociadas a síndrome de Cogan como ACV, encefalitis, meningitis asépticas, encefalopatía, trombosis venosa cerebral, compromiso de pares craneales, miopatías y neuropatías periféricas, sin embargo en mucho de estos casos no se encuentra evidencia suficiente para relacionarlos6. En cuanto al ACV, la mayoría de los casos publicados el diagnóstico del evento vascular fue definido por la clínica sin confirmación por imágenes ni haber descartado otras etiologías, por lo cual no existe gran evidencia de asociación entre estas patologías. Se cree que en caso de que exista, esta relación podría ser mediada por la vasculitis6.
Si bien existen criterios para el diagnóstico de esta enfermedad, estos no se encuentran validados5. Dichos criterios dividieron al síndrome de Cogan en típico y atípico, definiendo a este primero como aquellos pacientes con enfermedad ocular típica y síntomas audiovestibulares similares al Ménière con un intervalo entre ambas afecciones menor a dos años. Para aquellos pacientes que no cumplían estos criterios se los clasificó con síndrome de Cogan atípico, siendo más frecuente el compromiso sistémico en este último grupo8. En el laboratorio se suelen encontrar marcadores inflamatorios elevados (eritrosedimentación en 70%), anemia (15% de los casos), leucocitosis (36%) y trombocitosis (12%) y el estudio reumatológico y de la coagulación suelen resultar normales (89% de los pacientes)5,8. La RM podría ser útil, ya que estos pacientes tendrían la posibilidad presentar señales inflamatorias en el laberinto. Por su parte, el PET podría ser de utilidad como medio de diagnóstico para evaluar el compromiso de grandes vasos y permitir un diagnóstico precoz de vasculitis sistémicas para así aplicar un régimen de tratamiento más agresivo5.
El pronóstico es variable, siendo peor en pacientes con compromiso sistémico. En pacientes sin este, la sordera es la complicación más frecuente en hasta 50% de los sujetos5, en general irreversible7. Por el contrario, aquellos pacientes con compromiso sistémico, principalmente mediado por vasculitis, el pronóstico suele ser ominoso5.
No existen recomendaciones con adecuado nivel de evidencia con respecto a su tratamiento. La recomendación inicial es la administración de corticosteroides, con evidencia de mejoría de la audición y de los síntomas visuales9. En caso de ausencia de respuesta al tratamiento con corticosteroides, se plantea como segunda y tercera línea la utilización de inmunosupresores (ciclosporina, azatioprina, metotrexate) y terapia biológica, respectivamente7. La agresividad del tratamiento inicial va a variar según si el paciente presente o no compromiso sistémico5, existiendo poca información en estos últimos. Cuando hay disfunción vestibular es importante un rápido inicio de corticoides sistémicos para evitar la progresión a sordera. En aquellos pacientes con vasculitis se recomienda iniciar con corticoides sistémicos agregando, en casos de mala evolución, inmunosupresores como ciclofosfamida, metotrexate, entre otros8. Solo algunos reportes de casos han descrito la terapia biológica como tercera línea de tratamiento tras la falla de corticoides e inmunosupresores clásicos7. Los agentes biológicos probados fueron infliximab, etanercept, adalimumab y rituximab7. En general el resultado medido fue la pérdida de audición y no la evolución del compromiso sistémico. Si bien no hay evidencia suficiente, la experiencia clínica de estos fármacos en el síndrome de Cogan parece ser prometedora7.
El síndrome de Cogan es una entidad sumamente infrecuente que en un inicio fue descrita con compromiso exclusivo ocular y audiovestibular y que hoy es considerada una enfermedad sistémica. El diagnóstico es clínico y la utilidad de herramientas diagnósticas complementarias como el PET no está bien establecida, aunque podrían ser de ayuda para un diagnóstico precoz. La rareza de la enfermedad y, por ende, la falta de estudios prospectivos y randomizados hacen que la evidencia respecto a alternativas diagnósticas y terapéuticas sea limitada condicionando, en casos como el nuestro, una evolución desfavorable. Por esto es importante destacar que ante eventos vasculares recurrentes o de presentación inhabitual, no se deben olvidar las patologías inflamatorias locales o sistémicas, ya que un tratamiento oportuno podría mejorar el pronóstico.
Conflicto de interesesNinguno para declarar.
FinanciaciónNinguna.
Integrantes de la residencia de neurología del Hospital Italiano de Buenos Aires: Dr. María Cristina Zurru. Jefa de la Sección de Enfermedades Cerebrovasculares. Hospital Italiano de Buenos Aires, Argentina. Dr. Jorge Omar Norscini. Jefe de la Sección de Neuro-oftalmología. Hospital Italiano de Buenos Aires, Argentina.
