



The term camptocormia refers to marked flexion of the thoracolumbar spine, which resolves with supine positions, in the absence of fixed deformity.1 This symptom may have multiple aetiologies, including parkinsonian syndromes, paraspinal myopathies, dystonia, motor neuron diseases, and functional disorders.1–3 In exceptional cases, it can also be associated with POLG mutations.
We present the case of a 52-year-old man with history of mild psychomotor retardation since childhood; his father had late-onset Parkinson’s disease and his mother had essential tremor. His 2 older siblings were asymptomatic. The patient was referred to our department due to progressive anteroflexion of the trunk of 4 years’ progression, which hindered walking and was associated with poor coordination and slow movement, mainly affecting the right side; these symptoms were highly disabling. He displayed no signs of dysautonomia, with the exception of constipation.
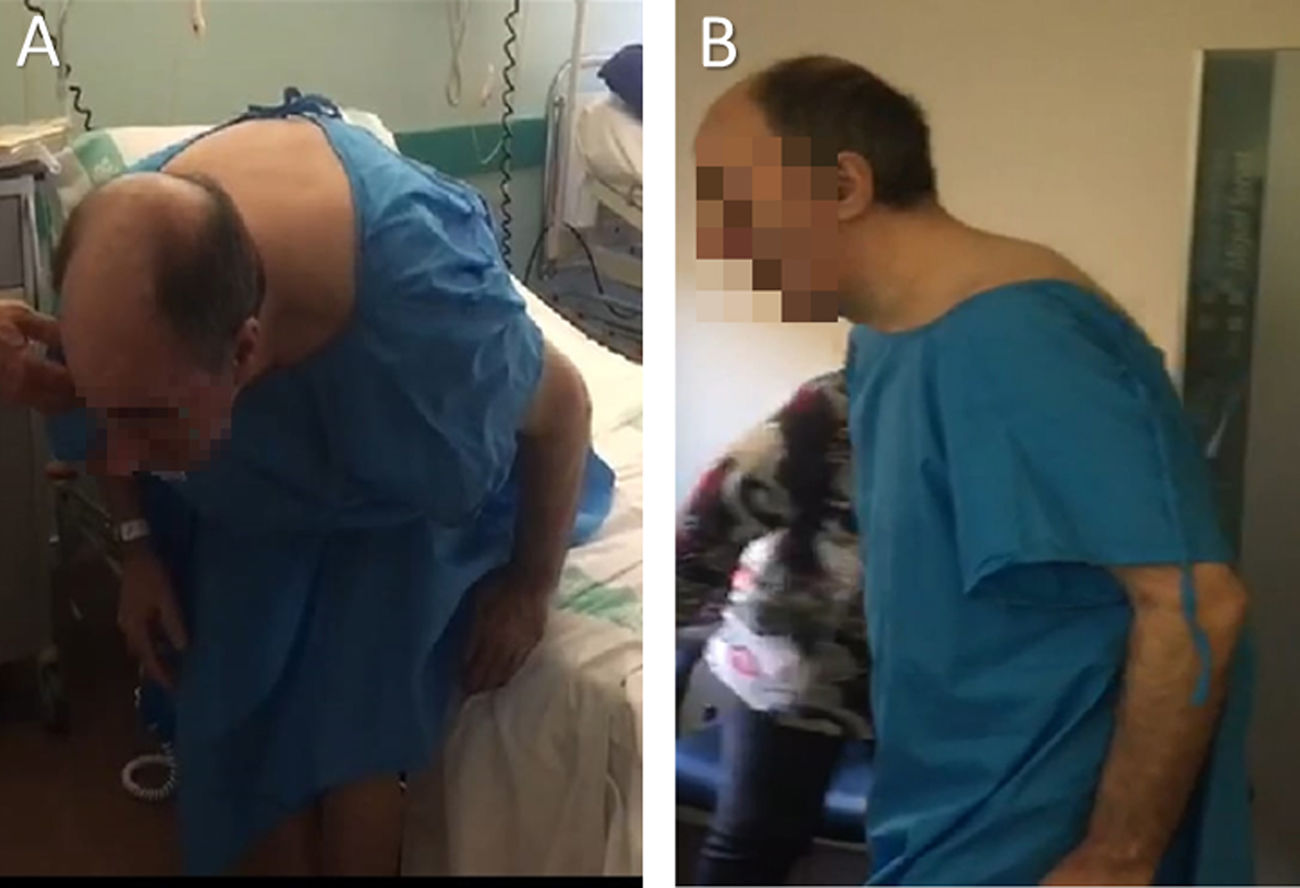
Neurological examination revealed hypomimia, moderate bilateral bradykinesia, mainly affecting the right side, and low-amplitude right-sided resting tremor. Eye movement was not restricted. When standing, the patient displayed forced anteroflexion of the trunk, with the waist at an angle of approximately 70° (Fig. 1). In the decubitus position, he was able to correct his posture, lying flat on the bed.
 and one hour after (B) a 350 mg dose of levodopa, showing a clear improvement in the anteroflexion of the trunk.")
A complete blood analysis including copper metabolism, muscle enzymes, and lactate detected no abnormalities; serology results for syphilis and HIV were negative. A brain MRI study detected no relevant alterations. A DaTSCAN study revealed dopamine transporter inactivity in both putamina and reduced uptake in the left caudate nucleus.
In the light of these findings, treatment was started with levodopa at increasing doses of up to 250 mg/6 hours, with a suboptimal response. After a 350 mg loading dose, he presented an improvement of over 30% on the UPDRS part III motor examination, with his posture improving and stabilising. The extreme anteroflexion of the trunk (Fig. 1) resolved, and he was able to carefully walk over 20 metres unassisted.
Given the history of psychomotor delay and the atypical parkinsonian symptoms, we requested an exome sequencing study, which identified a heterozygous missense mutation in exon 20 of the POLG gene (C.3218c > T; p [Pro1073Leu]), located on chromosome 15. The mutation is listed on the ClinVar and HGMD databases as a pathogenic variant. Functional studies have shown that this variant also affects mitochondrial DNA replication.4
Despite the lack of clear symptoms, we performed further diagnostic testing. An electromyoneurography study yielded normal results, and a multidisciplinary evaluation identified mild bilateral cataracts and predominantly sensorineural mixed hearing loss.
The POLG gene encodes the catalytic subunit of DNA polymerase gamma, which is responsible for the replication of the mitochondrial genome.5 Mutations of the gene are associated with a broad spectrum of neurological syndromes, with age of onset ranging from infancy to adulthood; manifestations include epilepsy, psychiatric disorders, polyneuropathy, myopathy, ataxia, and progressive external ophthalmoplegia, and may be associated with such non-neurological conditions as cataracts, sensorineural hearing loss, and premature ovarian failure.5,6
More rarely, POLG mutations may be associated with parkinsonism,7 often preceded for several years by classical phenotypes: usually progressive external ophthalmoplegia,8 but also sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO).9 In these patients, parkinsonism tends to be asymmetrical, with onset occurring around the age of 40 years and good response to levodopa.8,10 Our patient developed an atypical parkinsonian syndrome, with no evidence of other neurological characteristics associated with POLG mutations; prominent camptocormia was the most striking symptom. Additional tests performed after diagnosis identified bilateral cataracts and mild sensorineural hearing loss; these findings are associated with the mutation. This form of presentation, in which the parkinsonian syndrome is not preceded by ophthalmoparesis, has only been described in 2 patients also presenting peripheral neuropathy11 and in a patient with another pathogenic mutation in the GBA gene.12 We believe that the cause of camptocormia in our patient was parkinsonism itself, as he presented severe anteroflexion of the trunk and absence of abnormal postures in the head and neck (as would be the case for dystonic camptocormia), and posture improved (albeit suboptimally) with levodopa. Furthermore, the electromyography study showed no myopathic changes.
In conclusion, our patient presented a unique phenotype of POLG mutation, presenting with camptocormia secondary to atypical parkinsonism not preceded by SANDO or progressive external ophthalmoplegia, which has rarely been described in the literature.13POLG mutations should be considered in the differential diagnosis of atypical parkinsonism, even in patients without ophthalmoparesis or polyneuropathy.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Sancho Saldaña A, et al. Camptocormia como principal manifestación de mutación en el gen POLG. Neurología. 2021;36:390–392.
