




Alemtuzumab es un fármaco de alta eficacia aprobado por la Agencia Europea de Medicamentos como tratamiento modificador de la enfermedad en pacientes con esclerosis múltiple remitente recurrente.
ObjetivoElaborar un documento de consenso sobre el manejo de alemtuzumab en la práctica clínica habitual, que sea de aplicación en el ámbito español.
DesarrolloUn grupo de expertos en esclerosis múltiple revisó las publicaciones disponibles hasta diciembre de 2017, de tratamiento con alemtuzumab y esclerosis múltiple. Se incluyeron trabajos sobre eficacia, efectividad y seguridad, despistaje de infecciones y vacunación, administración y monitorización. La propuesta inicial de recomendaciones fue desarrollada por un grupo coordinador con base en la evidencia disponible y en su experiencia clínica. El proceso de consenso se llevó a cabo en 2 etapas; se estableció como porcentaje inicial de acuerdo grupal el 80%. El documento final con todas las recomendaciones acordadas por el grupo de trabajo se sometió a revisión externa y los comentarios recibidos fueron considerados por el grupo coordinador.
ConclusionesEl documento aportado pretende ser una herramienta útil para facilitar el manejo del fármaco en condiciones de práctica clínica habitual.
Alemtuzumab is a highly effective drug approved by the European Medicines Agency as a disease-modifying drug for the treatment of relapsing-remitting multiple sclerosis.
ObjectiveA consensus document was drafted on the management of alemtuzumab in routine clinical practice in Spain.
DevelopmentA group of multiple sclerosis specialists reviewed articles addressing treatment with alemtuzumab in patients with multiple sclerosis and published before December 2017. The included studies assessed the drug's efficacy, effectiveness, and safety; screening for infections and vaccination; and administration and monitoring aspects. The initial proposed recommendations were developed by a coordinating group and based on the available evidence and their clinical experience. The consensus process was carried out in 2 stages, with the initial threshold percentage for group agreement established at 80%. The final document with all the recommendations agreed by the working group was submitted for external review and the comments received were considered by the coordinating group.
ConclusionThe present document is intended to be used as a tool for optimising the management of alemtuzumab in routine clinical practice.
El aumento de opciones terapéuticas en esclerosis múltiple (EM) y su ascendente nivel de eficacia implican un incremento en la complejidad del manejo de aquellas, por lo que es necesaria la elaboración de documentos de consenso que uniformicen su uso y permitan una adecuada interpretación de su efectividad, monitorización y balance beneficio/riesgo.
Alemtuzumab es un anticuerpo monoclonal recombinante humanizado aprobado por la Agencia Europea de Medicamentos en septiembre del 2013 para el tratamiento de la esclerosis múltiple remitente recurrente (EMRR) como fármaco modificador del curso de la enfermedad1 (fecha de comercialización: marzo de 2015).
La incorporación de alemtuzumab como fármaco de alta eficacia en la EM, su innovador modo de acción y administración, junto con el creciente y amplio manejo que precisa han derivado en la necesidad de realizar un profundo análisis en relación con su uso. Hasta la fecha, varios autores han publicado recomendaciones y guías sobre aspectos concretos del manejo de alemtuzumab2-5.
El objetivo principal del presente consenso es profundizar en el manejo práctico de alemtuzumab, en términos relacionados con la efectividad, tolerabilidad y manejo de los posibles efectos adversos, así como con la selección del tipo de pacientes candidatos. Este documento incluye recomendaciones basadas en la evidencia procedente de estudios observacionales, análisis de series de pacientes y recomendaciones de los expertos.
Mecanismo de acciónAlemtuzumab es un anticuerpo monoclonal humanizado que reconoce CD52, una molécula que se expresa abundantemente en los linfocitos T y B maduros, pero no en sus precursores, y que apenas se expresa en las células de la respuesta innata como granulocitos6,7.
Alemtuzumab se une a las moléculas CD52 presentes en la membrana de los linfocitos circulantes en sangre periférica e induce su lisis por acción del complemento o por citotoxicidad mediada por anticuerpos. Esto induce una intensa linfopenia en sangre, con menor efecto en órganos linfoides secundarios, como los ganglios linfáticos o el bazo, y sin apenas influencia en la médula ósea8. La preservación de los progenitores linfoides hace posible la reconstitución de las poblaciones linfocitarias hemáticas. Durante dicha reconstitución se produce una reprogramación de la respuesta inmunitaria nueva, originándose un aumento en la ratio de células reguladoras frente a las efectoras, un claro aumento de la producción de citocinas antiinflamatorias y una disminución de las proinflamatorias9,10. Esto induce, en un alto número de pacientes, una estabilización prolongada o una mejoría de la enfermedad, aunque en ocasiones favorece la aparición de nueva enfermedad autoinmune. La preservación de la respuesta innata y el hecho de que los linfocitos no eliminados por el tratamiento11 sean plenamente funcionales podrían relacionarse con la baja tasa de infecciones graves que aparecen tras el tratamiento con alemtuzumab.
En resumen, alemtuzumab produce una reducción de los linfocitos circulantes y una posterior repoblación, con un cambio de la respuesta inflamatoria patológica hacia un fenotipo más tolerogénico, lo que origina en un alto porcentaje de los pacientes una remisión prolongada de la enfermedad.
Desarrollo clínico de alemtuzumabLa eficacia y seguridad de alemtuzumab (12mg) se han evaluado en 3ensayos clínicos frente a interferón ß-1a (44mg) subcutáneo en pacientes con EMRR activa. Los 2primeros ensayos incluían a pacientes no tratados previamente (estudio CAMMS223, fase ii12, 3años de duración y estudio CARE-MS I, fase iii13, 2años de duración), mientras que el tercero se realizó en pacientes con una respuesta inadecuada al tratamiento previo (estudio CARE-MS II, fase III, 2años de duración)14.
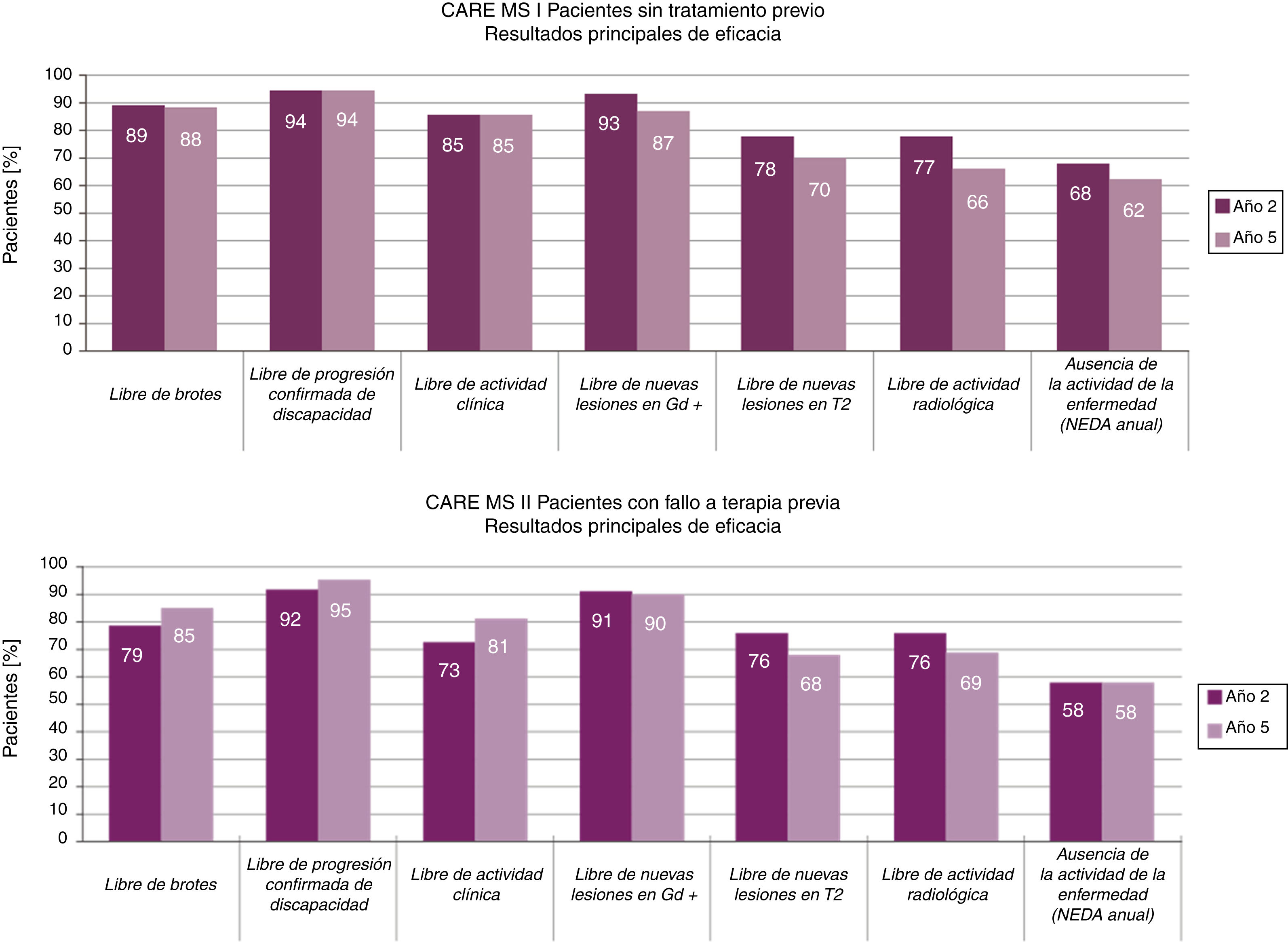
Eficacia de alemtuzumabEficacia en los ensayos aleatorizadosEn estos 3estudios el grupo tratado con 12mg de alemtuzumab presentó una reducción significativa del riesgo de brotes en un 69%12, 55%13 y un 49%14, respectivamente (p<0,001), así como una reducción significativa del riesgo de progresión confirmada de la discapacidad (cocientes de riesgos 0,25; p<0,00112 y 0,58; p=0,008)12,14 comparado con interferón ß-1a (el estudio CARE-MS I mostró un cociente de riesgo de 0,70; p=0,22)13. La tabla A.1 del anexo A muestra los resultados principales de los resultados clínicos y de resonancia magnética (RM) de los 2estudios principales CARE-MS13,14.
Eficacia a largo plazo en los estudios de extensiónEn los estudios de extensión CARE-MS (5 años de seguimiento) la tasa de retención de pacientes fue del 96%15 y del 91%16 desde el inicio de la fase de extensión, y del 89%15 y del 82%16 desde el inicio de los estudios principales17. Estas tasas de retención proporcionan gran solidez a los datos de eficacia de alemtuzumab y contrastan con las observadas en estudios a largo plazo de otros tratamientos modificadores de la enfermedad (TME)18-20.
La reducción de la tasa anualizada de brotes se mantuvo a lo largo del tiempo y pasó de 0,1813 y 0,2814 al finalizar los estudios principales (año 2, tasa anualizada acumulada los años 0-2) a 0,1615 y 0,2116 en los estudios de extensión (año 5, tasa anualizada acumulada los años 3-5) (fig. 1).

Tras 5 años, un 94-95%15,16 de los pacientes se mantuvo libre de progresión confirmada de la discapacidad y un 33-43%15,16 mostró una mejora confirmada de la discapacidad, durante un periodo de 6 meses. Los datos mostraron un aumento en la proporción de pacientes con mejora confirmada del año 2 al 5 (en los años 2, 3, 4 y 5, MS-CARE I: 24, 28, 30 y 33%; MS-CARE II: 29, 35, 41 y 43%)15,16.
El examen con RM (imágenes potenciadas en T2 y en T1 con gadolinio) en los estudios de extensión mostró una ausencia de nuevas lesiones o empeoramiento de las previas en más del 75% de los casos, en todos los estudios15,16 (fig. 1).
El porcentaje anualizado de pacientes en estado NEDA (ausencia de actividad de la enfermedad, acrónimo del inglés no evidence of disease activity, que incluye ausencia de actividad clínica —sin brotes ni progresión de la discapacidad— y radiológica —RM sin lesiones con captación de contraste ni lesiones de nueva aparición/aumento de tamaño en secuencia T2—) se mantuvo estable alrededor del 60% a lo largo de los estudios de extensión15,16.
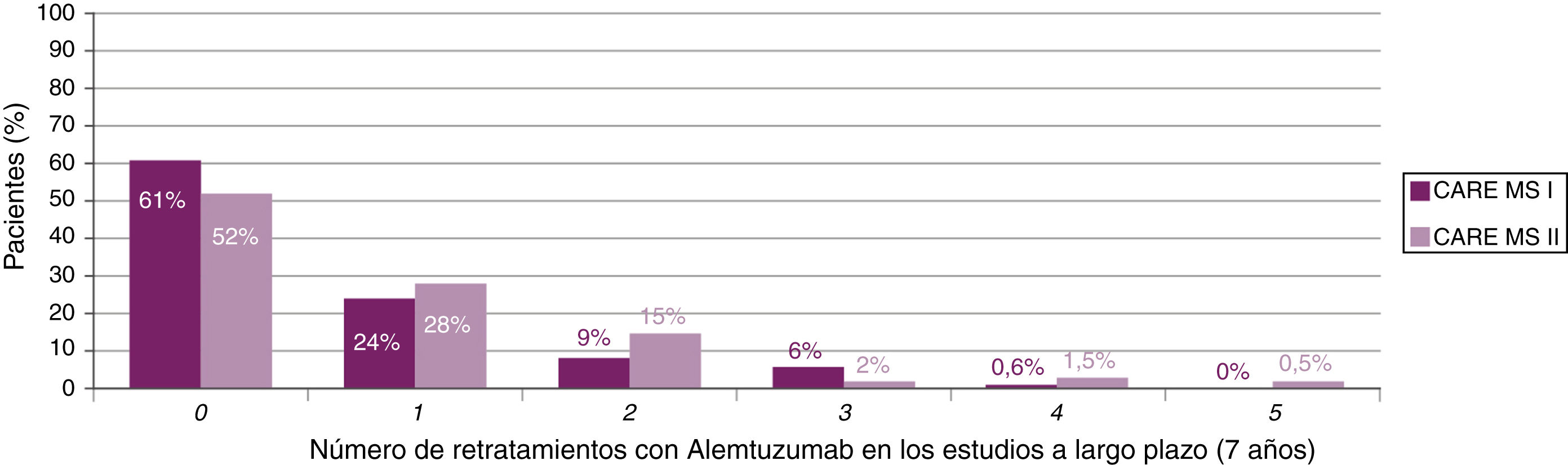
Los resultados del estudio TOPAZ (extensión conjunta de MS-CARE I y II tras 7 años de seguimiento) presentados en el 7th Joint ECTRIMS-ACTRIMS Meeting consolidan los resultados previos de eficacia en ausencia de tratamiento continuado, tanto en pacientes sin tratamiento previo como en no respondedores al TME, con una reducción sostenida de la actividad de RM y de la pérdida de volumen cerebral21,22, así como una mejora de las variables clínicas23,24.
La mayoría de los pacientes no recibió cursos adicionales después del segundo curso de alemtuzumab (fig. 2). En el caso de requerirlo, los motivos durante 7 años fueron (para MS-CARE I y II, respectivamente): brotes (50%23 y 57%24), actividad radiológica (28%23 y 19%24) o ambos (20%23 y 21%24).

Se han descrito claros beneficios a largo plazo tras la administración del segundo curso de alemtuzumab en pacientes que presentan brotes entre el primero y el segundo curso25,26.
Asimismo, existen nuevos datos que confirman un claro beneficio de un tercer curso de alemtuzumab en pacientes que presenten actividad de la enfermedad tras los 2primeros cursos25,27.
Seguridad de alemtuzumabSeguridad en los ensayos aleatorizadosAlemtuzumab presenta un buen perfil de seguridad, con un efectivo protocolo de detección y tratamiento de sus efectos adversos.
En los estudios CAMMS223, CARE-MS I y II se observaron principalmente reacciones asociadas a la perfusión (más del 90%), infecciones (66, 67 y 77%, respectivamente) y enfermedades autoinmunes secundarias: un 26, 16 y 18% de trastornos tiroideos; un 1,9; 1 y 1% de púrpura trombocitopénica inmune y un 0,3% de nefropatías en los 3estudios agrupados12-14.
Seguridad a largo plazo en los estudios de extensiónEl perfil de seguridad de alemtuzumab es consistente, con una incidencia anual de eventos adversos que se reduce tras el primer año de tratamiento.
La exposure-adjusted incidence rate para la totalidad de los eventos adversos fue más baja en los estudios de extensión que en los principales15,16.
La mayoría de los eventos adversos fueron de intensidad leve o moderada y no hubo retiradas en los estudios debido a estos. Ninguno de los eventos adversos relacionados con la perfusión fue grave. Las principales reacciones asociadas a la perfusión fueron cefalea, pirexia y erupciones cutáneas. La incidencia de infecciones tampoco aumentó y la mayoría fueron leves o moderadas (99,2 y 97,7%). Las más frecuentes fueron nasofaringitis, infecciones del tracto urinario, infecciones del tracto respiratorio superior e infecciones por herpes15,16.
Los eventos adversos autoinmunes más comunes fueron los trastornos tiroideos, con un pico de incidencia mayor al tercer año (16,7 y 16,5%, datos de los estudios de extensión15,16).
Los datos de seguridad tras 7 años de seguimiento muestran una reducción de la frecuencia de eventos adversos respecto a los estudios principales. El séptimo año, la incidencia de trastornos tiroideos e infecciones fue del 1,6 y 30,8% (frente al 6,4 y 56,1% del primer año, respectivamente23). Los resultados son similares en pacientes previamente no respondedores a los TME (CARE-MS II)24.
En la extensión del estudio CAMMS223, tras 10 años de seguimiento, alemtuzumab muestra un perfil de seguridad consistente con el estudio principal28.
Desarrollo del consensoPara la formulación de las recomendaciones se ha utilizado una metodología de consenso de tipo formal (modified nominal group technique29). El proceso de consenso se llevó a cabo en 2etapas y se estableció a priori como punto de corte para aceptar una recomendación un 80% de acuerdo en el grupo.
En la primera etapa, los participantes recibieron por correo electrónico un resumen de la evidencia disponible sobre los aspectos priorizados (eficacia, efectividad y seguridad, despistaje de infecciones y vacunación, administración y monitorización) y una propuesta de recomendaciones elaboradas por el panel multidisciplinar con base en la evidencia disponible y en su experiencia clínica. Además, se envió también una explicación general de la metodología y una hoja de clasificación del grado de acuerdo, utilizando una escala de 9 puntos de tipo Likert: 1-3 (estrategia inapropiada), 4-6 (incierta) y 7-9 (estrategia apropiada) (anexo B). En esta fase, las recomendaciones fueron sometidas a una primera ronda de votación individual por los participantes en el consenso (grupo de trabajo) utilizando la escala de puntos de tipo Likert anteriormente mencionada. El grupo de trabajo tenía la posibilidad de añadir nuevas recomendaciones o modificar las ya propuestas si estaban en desacuerdo.
Después, el grupo de trabajo recibió por correo electrónico un resumen de la evidencia disponible sobre las recomendaciones propuestas por el grupo coordinador y los resultados del grado de acuerdo en la primera votación (anexo B).
En una segunda etapa, el grupo coordinador y el de trabajo celebraron una reunión de consenso presencial, conducida por 2moderadores miembros del grupo coordinador (XM y OF). Se presentó de forma anonimizada el resultado de la primera votación individual. Las recomendaciones con menos del 80% de acuerdo se volvieron a redactar y se llevó a cabo una segunda votación a mano alzada. Aquellas recomendaciones que no pudieron ser aprobadas durante la reunión presencial debido a limitaciones de tiempo fueron consensuadas posteriormente vía correo electrónico. Finalmente, las recomendaciones que no consiguieron la aprobación en esta fase no fueron incluidas en el consenso.
RecomendacionesSelección del paciente candidato a tratamiento con alemtuzumabSegún la ficha técnica30, alemtuzumab puede ser utilizado en pacientes adultos con EMRR con enfermedad activa definida por manifestaciones clínicas o detectadas por RM. En el estudio CARE-MS II, la mayoría de los TME previos fueron interferón β-1a y acetato de glatirámero, natalizumab solo representó el 3% y fingolimod no estaba aprobado en el momento en que se realizó el estudio14. Por lo tanto, la evidencia sobre el manejo de pacientes que reciben alemtuzumab después de tratamientos con fingolimod, natalizumab y otros inmunosupresores proviene principalmente de las publicaciones de series de pacientes tratados en la práctica clínica real31.
Recomendación 1Si se integran las recomendaciones de la ficha técnica y el perfil clínico basal de los pacientes incluidos en los ensayos clínicos CARE-MS II y II, se podría concluir que las características del paciente candidato a alemtuzumab deberían aproximarse a las siguientes: enfermedad activa clínica o radiológica o respuesta subóptima a un curso completo de TME o enfermedad con curso rápido y agresivo.
Recomendación 2La utilización de alemtuzumab en pacientes con EM tratados previamente con TME inmunosupresores selectivos no se ha asociado en la práctica clínica diaria con problemas de seguridad en las series descritas hasta la fecha. Se recomienda adoptar medidas de seguridad adicionales a lo establecido en la ficha técnica en aquellos pacientes que van a iniciar tratamiento con alemtuzumab y que previamente han sido tratados con otros fármacos inmunosupresores.
Consideraciones previas al inicio del tratamientoInfecciones asociadas al uso de alemtuzumabEn los estudios CARE-MS I y II no se produjo un aumento en la incidencia de infecciones graves ni oportunistas. No obstante, en estos ensayos clínicos principales se describieron pacientes con tuberculosis activa (0,3%), fundamentalmente procedentes de áreas endémicas. En la práctica clínica se han descrito casos aislados de listeriosis32,33, nocardiosis del sistema nervioso central34, neumonitis30 y neumonitis asociada con pericarditis35. No se han descrito casos de leucoencefalopatía multifocal progresiva. No se dispone de datos de potencial reactivación de infecciones crónicas por virus de la hepatitis B o C debido a que estos pacientes fueron excluidos de los ensayos clínicos.
No se han identificado factores de riesgo específicos para el desarrollo de infecciones en los pacientes con EMRR tratados con alemtuzumab. Aunque existe una incidencia algo mayor en el primer mes tras la administración del fármaco, las infecciones se distribuyen uniformemente a lo largo de los meses de seguimiento y tienden a disminuir con el tiempo36. No se ha encontrado relación entre la incidencia de infecciones y el grado de linfopenia (absoluta y de linfocitos T CD4+)37.
Infecciones prevenibles por vacunaciónLa vacunación en pacientes con EM en tratamiento inmunosupresor o inmunomodulador forma parte del abordaje multidisciplinar de esta entidad. El efecto de estos fármacos sobre el sistema inmune puede aumentar el riesgo de infecciones, como encefalitis y meningitis causadas por los virus herpes simple y varicela zóster38,39.
Desde el punto de vista legal, la indicación de vacunación en estos pacientes siguiendo las recomendaciones oficiales de vacunación no precisa consentimiento informado por escrito.
Recomendación 3Se recomienda abordar de manera temprana en el curso de la enfermedad la vacunación del paciente con EM para asegurar una mayor respuesta a las vacunas. Se ha demostrado, por ejemplo, que en el caso de la vacunación frente a neumococo la inmunogenicidad es menor cuando el paciente recibe inmunosupresores en monoterapia40. Como norma general, las vacunas vivas-atenuadas están contraindicadas en embarazadas y en pacientes en tratamiento inmunosupresor. Por el contrario, no existe contraindicación para la vacunación con vacunas inactivadas durante la terapia con TME41, aunque es importante valorar el mecanismo de acción del fármaco TME en busca del momento óptimo de vacunación, con el objetivo de conseguir el mayor efecto protector.
Estrategias de vacunación con alemtuzumabRecomendación 4Se aconseja evaluar la necesidad de vacunación antes de iniciar tratamiento con alemtuzumab. Para ello es necesario: a) conocer el estado serológico del paciente frente a sarampión, varicela, hepatitis A y hepatitis B; b) documentar los antecedentes de vacunación del paciente, con especial énfasis en gripe estacional, neumococo, virus de la hepatitis A y B, tétanos-difteria, varicela, sarampión y virus del papiloma humano.
Recomendación 5Se aconseja evaluar la vacunación en pacientes que van a iniciar alemtuzumab según las especificaciones de la tabla 1. En ella se describen las vacunas que valorar, su composición y la pauta de primovacunación y dosis de recuerdo.
Vacunas valorables en el paciente que va a recibir alemtuzumab48,49
| Vacuna | Tipo | Indicaciones | Pauta | Dosis de recuerdo |
| Gripe estacional | Inactivada.Fraccionada o de subunidades | Sí | 1 dosis anual en campaña de vacunación antigripal (octubre-diciembre) | No precisa |
| Neumococo 13v | Inactivada.Polisacárido conjugado con proteína | Sí | 1 dosis | No precisa |
| Neumococo 23v | Inactivada. Polisacárido | Sí | 1 dosisA partir de los 2 meses de la administración de la vacuna antineumocócica conjugada 13v | A los 5 años |
| Hepatitis Ba | Inactivada.Antígeno de superficie | Sí, siempre y cuando antiHBs <10 UI/mL | 0-1-2-6 mesesd | No precisa |
| Hepatitis A | Inactivada. Virus enteros | Sí, siempre y cuando IgG VHA (-) | 0-6 meses | No precisa |
| Tétanos-difteria | Inactivada. Toxoide tetánico | Sí. Deberá iniciarse pauta de primovacunación si no la ha recibido en el pasado o completar con dosis de recuerdo | Primovacunación (3 dosis):0-1-6 meses o 0-1-12 meses | Dos dosis: la 1.ª deberá ser a los 10 años de la primovacunación y la 2.ª a los 10 años de la primera dosis de recuerdo |
| Virus del papiloma humano | Inactivada. Subunidades | Síb | 0-1-6 meses o 0-2-6 meses | No precisa |
| Varicelac | Viva-atenuada. Virus entero | Sí, siempre y cuando IgG varicela (−) | 0-1 mes | No precisa |
| Triple vírica(sarampión-rubeola-parotiditisc) | Viva-atenuada. Virus entero | Sí, siempre y cuando IgG sarampión (−) | 0-1 mes | No precisa |
antiHBs: anticuerpo de superficie de la hepatitis B; Ig: inmunoglobulina; VHA: virus de la hepatitis A.
Podrá considerarse la administración de vacuna adyuvada con AS04C, vacuna de alta carga VHB 40 mcg, o doble dosis de vacuna convencional manteniendo la misma pauta de vacunación.
Preferentemente en mujeres menores de 26 años de edad que no hayan recibido la vacuna siguiendo el calendario sistemático infantil. También en mujeres en las que se vaya a realizar tratamiento escisional de cérvix por lesiones de alto grado (HSIL/CIN 2-3). En este caso, administrar de forma precoz tras el diagnóstico de la lesión a ser posible antes de la intervención y hasta 12 meses tras ella.
En caso de estar indicada la vacunación con más de una vacuna viva-atenuada deberá llevarse a cabo en el mismo acto vacunal en distinto lugar anatómico. Si no se administran juntas, deberá respetarse un intervalo de, al menos, 4 semanas entre ambas vacunas.
Se recomienda realizar una serología postvacunal un mes después de la finalización de la pauta vacunal para confirmar una correcta respuesta vacunal (título antiHBs>10UI/mL). En los individuos que no seroconviertan se recomienda repetir la vacunación con 3 dosis adicionales a intervalos mensuales. La revacunación se realizará con vacuna adyuvada con AS04 o con vacuna con alta carga (40 mcg). Los pacientes que no hayan conseguido seroconvertir tras la repetición de la vacunación se considerarán «no respondedores» y deberá tenerse en cuenta la profilaxis postexposición en caso de situación de riesgo.
Los procesos de vacunación pueden extenderse entre más de 2 meses y un año, por lo que el clínico valorará el balance beneficio-riesgo. En caso de no poder retrasar el inicio de alemtuzumab se recomienda respetar los intervalos entre la vacunación y el inicio de tratamiento con alemtuzumab establecidos en la tabla 2.
Estrategia de vacunación cuando exista indicación de tratamiento con alemtuzumab
| Vacunación prealemtuzumab | Vacunas vivas-atenuadas | Vacunas inactivadas |
|---|---|---|
| No TME previo | Finalizar pauta de vacunación al menos 6 semanas antes de iniciar alemtuzumab | No contraindicación para la vacunación y no hay intervalo mínimo para el inicio del tratamiento, pero la respuesta pudiera ser menor de la esperada |
| Tratamiento esteroideo reciente (tratamiento de un brote) | Iniciar vacunación al menos 1 mes tras la última dosis en caso de prednisona> 20 mg/día (o equivalente) durante más de 15 díasSi el paciente ha recibido bolo de corticoides en los últimos 3 meses se recomienda valoración individual de la situación inmunológica | No contraindicación para iniciar vacunación, pero la respuesta pudiera ser menor de la esperada cuando recibe corticoides en dosis inmunosupresoras |
| Finalizar pauta de vacunación al menos 6 semanas antes de iniciar alemtuzumab | ||
| Tratamiento activo con interferón ß o acetato de glatirámero | No contraindicación para iniciar vacunación durante tratamiento con estos fármacos (salvo que se encuentren en combinación con inmunosupresores), pero la respuesta pudiera ser menor de la esperada | |
| Finalizar pauta de vacunación al menos 6 semanas antes de iniciar alemtuzumab | No contraindicación para iniciar vacunación, pero la respuesta pudiera ser menor de la esperada | |
| Tratamiento activo con teriflunomida o DMF | Iniciar vacunación una vez se haya completado el periodo de lavado (ver tabla 3)a | No contraindicación para iniciar vacunación, pero la respuesta pudiera ser menor de la esperada |
| Finalizar pauta de vacunación al menos 6 semanas antes de iniciar Alemtuzumab | ||
| Tratamiento activo con inmunosupresores específicos, natalizumab o fingolimod | Iniciar vacunación al menos 3 meses tras la última dosis de alguno de estos fármacosa | No contraindicación para iniciar vacunación, pero la respuesta pudiera ser menor de la esperada |
| Finalizar pauta de vacunación al menos 6 semanas antes de iniciar alemtuzumab | ||
| Tratamiento activo con alemtuzumab | Contraindicadas durante el tratamiento. Pasar a vacunación postalemtuzumab | No contraindicación para iniciar vacunación, pero la respuesta pudiera ser menor de la esperada. Serecomienda retrasar la vacunación (postalemtuzumab) para conseguir inmunización efectiva |
| Vacunación postalemtuzumab | Vacunas vivas-atenuadas | Vacunas inactivadas |
| Contraindicadas hasta pasados 6 meses y siempre y cuando se establezca la reconstitución inmunológica sistémicab | No contraindicación, pero la respuesta pudiera ser menor de la esperada. Esperar reconstitución inmunológica sistémicab | |
DMF: dimetilfumarato; TME: tratamiento modificador de la enfermedad.
Como el periodo ventana puede ser prolongado, valorar iniciar alemtuzumab y pasar al apartado vacunación postalemtuzumab. No obstante, no debería ser una limitación para iniciar alemtuzumab, ya que el calendario de vacunación se debería haber confirmado antes de iniciar interferón ß, acetato de glatirámero, teriflunomida, DMF, natalizumab o fingolimod. Valorar cada caso de forma particular.
- •
Deben excluirse infecciones activas mediante anamnesis, exploración física y pruebas complementarias adecuadas. La presencia de una infección activa obliga a su tratamiento adecuado y a retrasar el inicio del tratamiento con alemtuzumab hasta la curación.
- •
Se recomienda el despistaje de infección tuberculosa latente preferentemente mediante la prueba de ensayos de liberación de gamma-interferón específico (IGRA) o prueba de la tuberculina, siguiendo los protocolos de cada centro hospitalario. Los pacientes en los que se documente una infección tuberculosa deben recibir tratamiento con isoniacida (300mg/día en ayunas, durante 6 meses), suplementada con vitamina B6. El tratamiento de la infección tuberculosa no debe retrasar el inicio de tratamiento con alemtuzumab. Según las posibilidades, se recomienda retrasar 2-4 semanas para comprobar la tolerabilidad y ausencia de toxicidad de isoniacida.
- •
Despistaje de otras infecciones que ayuden a tomar decisiones de cara a la vacunación adecuada o al riesgo de complicaciones (hepatitis B, hepatitis C, virus de la inmunodeficiencia humana, citomegalovirus, varicela zóster, virus de papiloma humano; Strongyloides en inmigrantes de ciertas áreas endémicas como Hispanoamérica y norte de África).
- •
Prevención de infección herpética: durante los ensayos clínicos de fase III se demostró que el tratamiento con aciclovir previene el desarrollo de infecciones herpéticas42,43. Se debe administrar profilaxis oral con aciclovir desde la primera dosis de alemtuzumab (200mg/12h durante 4-8 semanas). Como alternativa, puede también administrarse valaciclovir (500mg/día) durante el mismo periodo.
- •
Minimización del riesgo de infección por listeria: se debe evitar la ingesta de alimentos cárnicos crudos o poco cocinados y lácteos no pasteurizados desde 2semanas antes del inicio del tratamiento y hasta como mínimo un mes después de la administración o hasta la reconstitución inmunológica sistémica, según los casos.
- •
Tras la administración de alemtuzumab no se recomienda la utilización sistemática de medidas preventivas específicas de infecciones bacterianas, fúngicas ni parasitarias. No obstante, ante síntomas sugestivos es necesario descartar infecciones graves, sean de carácter oportunista o no.
En este documento de consenso diferenciamos 3periodos durante el tratamiento con alemtuzumab: pretratamiento, tratamiento y postratamiento.
Pretratamiento- •
Informar al paciente sobre el balance beneficio/riesgo del tratamiento y la importancia del plan de seguimiento (plan de minimización de riesgos).
- •
Establecer indicaciones dietéticas de prevención de infección por listeria (ver recomendación 10).
- •
Firma del consentimiento informado.
- •
Evaluación de tratamientos previos (lavado) y concomitantes (tabla 3).
Tabla 3.Evaluación de tratamientos previos (lavado) y concomitantes
Otras recomendaciones Tiempo de espera Otras recomendaciones Sin tratamiento previoInterferón ßAcetato glatirámero No esperar -------- Teriflunomida ----------- Procedimiento de eliminación acelerada 11 días tras el cese del tratamiento:8g de colestiramina 3 veces al día vía oral (recomendado)o 50g de carbón activado 2 veces al día vía oralComprobar que los niveles sanguíneos son menores de 0,02 mg/L4 Dimetil fumarato ------------ Cuando el recuento linfocitario sea superior a 800 células/μL50 Fingolimod 6 semanas Siempre y cuando el recuento linfocitario sea superior a 800 células/μL51 Natalizumab 12 semanas JC (−), 12 semanas después de la última perfusión de natalizumab9 y realizar una RM cerebral Gd+antes de la perfusión de alemtuzumab para despistaje de LMPJC (+) el periodo ventana debería permitir descartar LMP latente antes de la perfusión de alemtuzumab, aunque la excesiva duración podría favorecer un rebrote (también denominado IRIS-like) por retirada de natalizumab. En estos casos debería plantearse un estudio del número de copias JC en LCR para descartar LMP latente39. También se debe realizar RM cerebral Gd+antes de la perfusión de alemtuzumab para despistaje de LMP, así como a los 6 meses y al año de la administración de alemtuzumab52 Otros inmunosupresores (azatioprina, ciclofosfamida, rituximab) Entre 3 y 6 meses Siempre que el recuento linfocitario sea>800células/μL y el recuento de CD4>200 células/μL Gd+: gadolinio; JC: virus John Cunningham; LCR: líquido cefalorraquídeo; LMP: leucoencefalopatía multifocal progresiva; RM: resonancia magnética.
- •
Análisis sanguíneo y urinario basal previo a la perfusión: hemograma, función renal, tiroidea, bioquímica y despistaje de proteinuria.
- •
Test de embarazo y medidas contraceptivas. Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces cuando reciban un curso de tratamiento con alemtuzumab y durante los 4 meses posteriores15,16.
- •
Despistaje de infecciones activas, prevenibles mediante vacunación y de infección tuberculosa latente (véase apartado «Consideraciones previas al inicio del tratamiento»).
- •
Protocolo de vacunación (anexo).
- •
Electrocardiograma y radiografía de tórax reciente. En un paciente asintomático puede ser válida una radiografía realizada en los 2 meses previos al día de la perfusión.
La dosis recomendada de alemtuzumab es de 12mg/día, administrados por perfusión intravenosa en 2 cursos de tratamiento inicial, con hasta 2 cursos adicionales de tratamiento, si fuera necesario, según establece la ficha técnica (1.er curso: 5 días de tratamiento; el 2.° y cursos sucesivos, 3 días consecutivos)30.
Recomendación 16- •
La administración de alemtuzumab puede realizarse mediante hospitalización interna y hospitalización ambulatoria u hospital de día. Estas opciones son válidas y la elección de una u otra dependerá de la infraestructura disponible en el hospital, del acceso a camas hospitalarias, de la disponibilidad de hospital de día de mañana y tarde, entre otros factores.
- •
De acuerdo con las recomendaciones recogidas en la sección sobre el riesgo de infección, antes de administrar alemtuzumab:
- o
Iniciar tratamiento con aciclovir (200mg/12h) y mantener durante un mínimo de 4-8 semanas.
- •
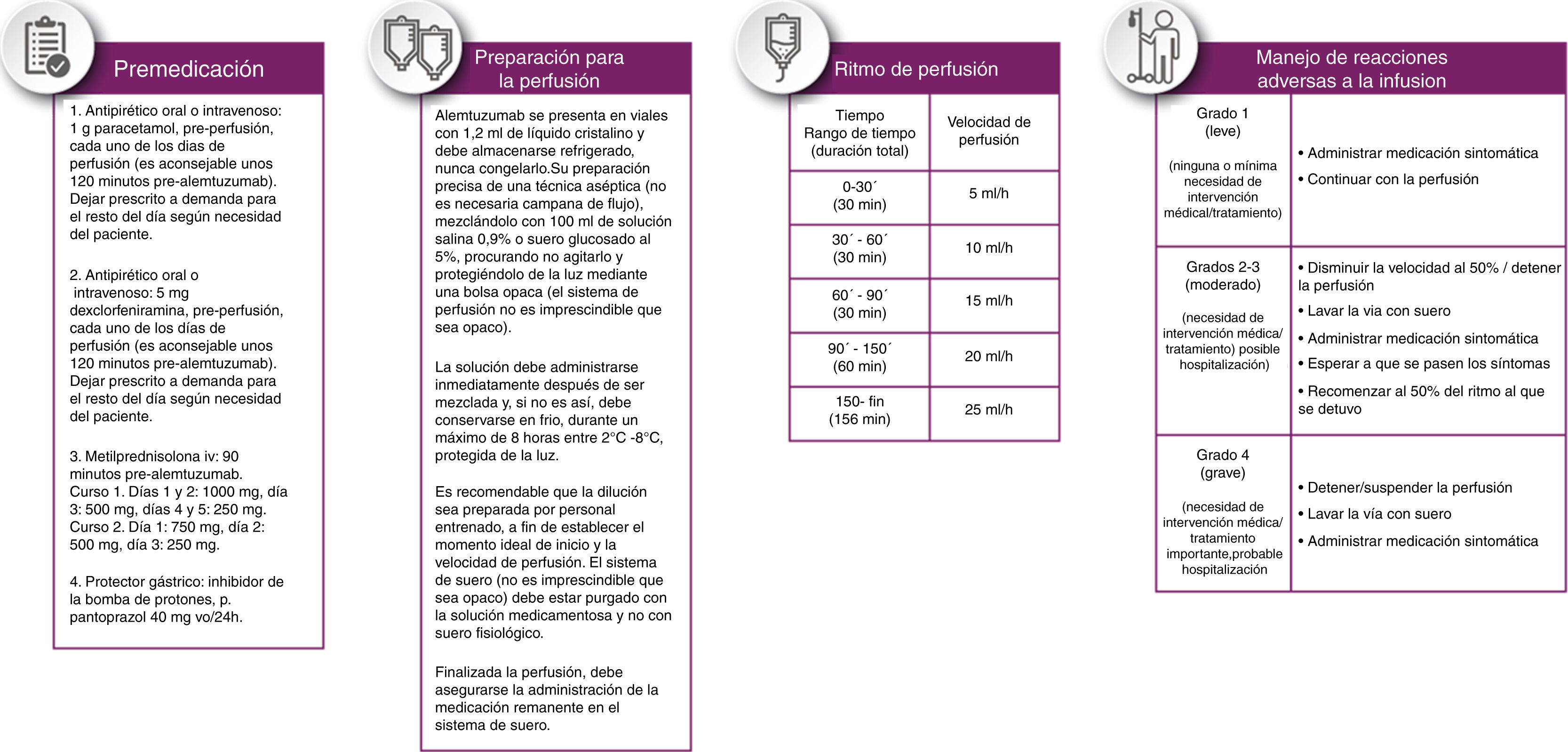
Se recomienda administrar premedicación a los pacientes para reducir la incidencia e intensidad de RAP (ver fig. 3 apartado de premedicación).
- •
El fármaco debe administrarse en no menos de 4 h ni más de 8 h con posibilidad de disminuir la velocidad si el paciente experimenta RAP. Se ha consensuado el ritmo de perfusión recogido en la figura 3.

Tras la perfusión del fármaco, los pacientes deben permanecer en observación durante 2 h sin retirar el acceso venoso periférico.
Monitorización durante la perfusión y manejo de las reacciones asociadas a la perfusión- a)
Monitorización y observación durante la perfusión y la posperfusión.
- •
Se recomienda realizar mediciones de frecuencia cardiaca, presión arterial y temperatura cada hora durante la perfusión y durante las 2h de observación tras la perfusión. Se pueden acompañar de inspecciones dermatológicas para detectar la presencia de urticaria, erupciones y otras lesiones dermatológicas o vasculares menos frecuentes.
- b)
Manejo de las reacciones asociadas a la perfusión
- •
Se recomienda abordar el tratamiento de las RAP en función de su gravedad siguiendo las indicaciones recogidas en la figura 3.
- •
Tratamiento sintomático oral domiciliario con antihistamínico y antipirético si aparecen síntomas como erupciones o cefalea. Indicar al paciente que debe volver al hospital si tras este tratamiento sintomático los síntomas persisten o aumentan.
- •
Proporcionar la tarjeta del paciente con la medicación administrada (plan de minimización de riesgos).
- •
Recomendaciones generales para disminuir el riesgo de infección: mantener una adecuada higiene corporal y de manos, y evitar ambientes poco ventilados y lugares en los que se produzca hacinamiento humano.
- •
Mantener indicaciones dietéticas.
- •
Análisis sanguíneo y urinario mensual durante los 48 meses posteriores al último curso: hemograma, función renal, función tiroidea trimestral, bioquímica y despistaje de proteinuria.
- •
Garantizar accesibilidad médica a otros especialistas (por ejemplo, endocrino, hematólogo, nefrólogo) para consultar sobre posibles eventos adversos.
- •
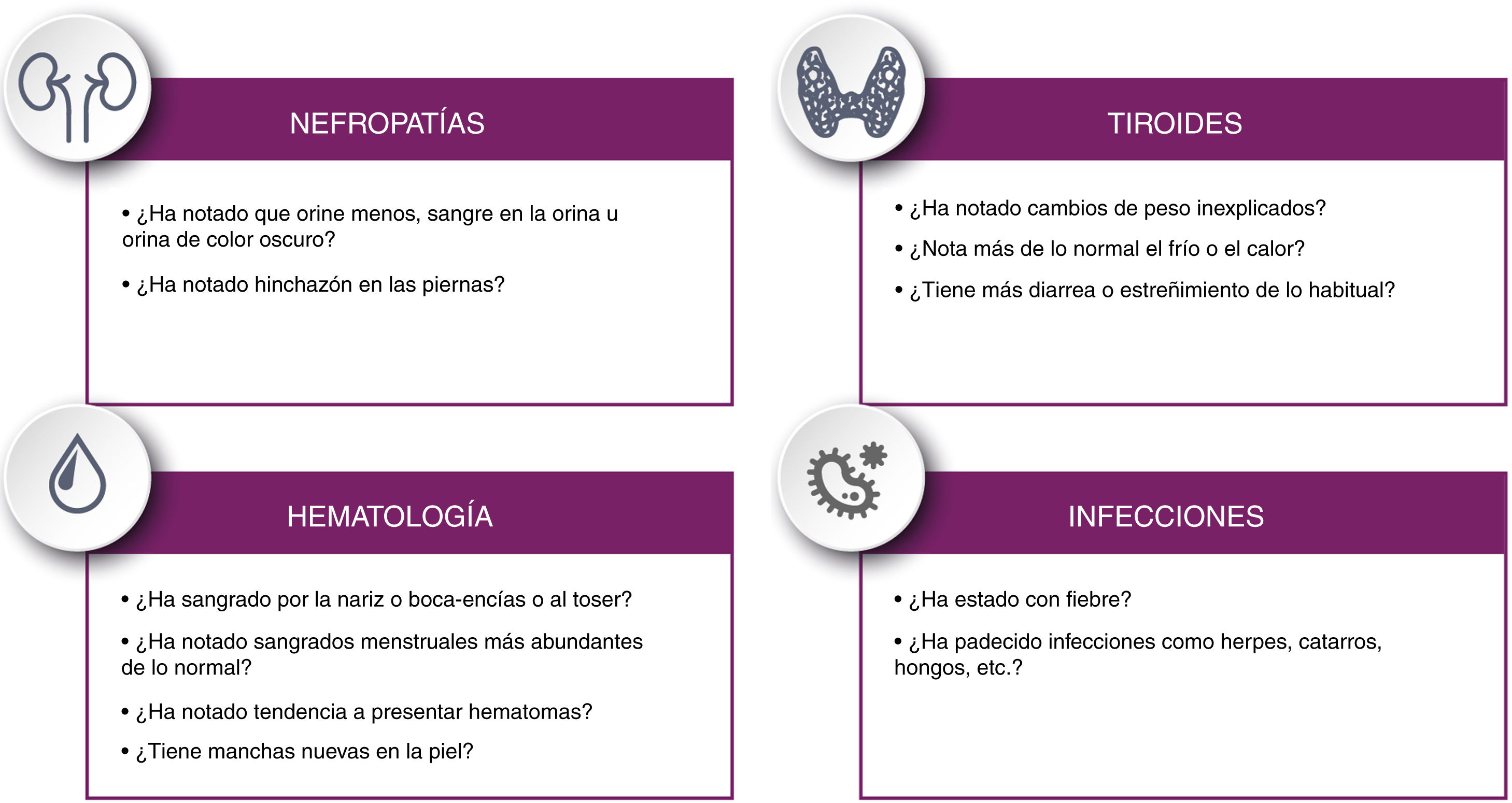
Se recomienda instruir al paciente sobre los síntomas que podría experimentar indicativos de las alteraciones inmunológicas que pueden aparecer a largo plazo siguiendo la lista de comprobación propuesta en la figura 4.

- •
Se recomienda una citología ginecológica con determinación del virus del papiloma humano anualmente en mujeres tratadas con alemtuzumab.
El resumen de los 3periodos se recoge en la figura 5.

Resumen de las recomendaciones durante los 3 periodos de administración de alemtuzumab.
CMV: citomegalovirus; RAP: reacciones asociadas a la perfusión; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; VIH: virus de inmunodeficiencia humana; VPH: virus del papiloma humano; VVZ: virus varicela-zóster.
- •
Deben trascurrir 12 meses entre el primer y segundo curso de alemtuzumab. Se recomienda alargar este periodo en caso de que el paciente no haya alcanzado una reconstitución inmunológica sistémica (recuento LT CD4+>200 cél/μL) o porque exista alguna complicación activa derivada del primer curso (por ejemplo, alguna alteración tiroidea).
En los ensayos clínicos, la administración del segundo curso ha mostrado eficacia en aquellos pacientes que presentaron brotes después del primero25,26. Los datos preliminares demuestran que el 42% de pacientes que han recibido alemtuzumab redujeron la tasa anualizada de brotes significativamente de 1,29 a 0,34 el primer año tras el segundo curso y no recibieron tratamiento adicional durante 6 años25,26.
Recomendación 24- •
En el segundo curso de alemtuzumab y posteriores, se recomienda la realización de los mismos procedimientos llevados a cabo durante los 3periodos establecidos para el primer curso.
En los estudios de extensión de los ensayos CARE-MS se permitió la administración de cursos adicionales de alemtuzumab 12 meses o más desde el último curso. Los criterios de eligibilidad fueron ≥1 brotes definido por protocolo o ≥ 2 nuevas/aumento lesiones hiperintensas en T2 o lesiones captantes de gadolinio en secuencia T1. La decisión sobre si iniciar el retratamiento en pacientes elegibles se dejó al médico y paciente, así como la decisión de proporcionar otra TEM autorizada15,16.
Un análisis en pacientes que recibieron un tercer curso de alemtuzumab mostró que la tasa anualizada de brotes se redujo de 0,74 en el año anterior al retratamiento hasta menos de 0,1 en los 3 años posteriores27. Estas cifras son similares a las de pacientes que no recibieron un curso adicional. Un año después del tercer curso, la puntuación media en la escala Expanded Disability Status Scale se redujo en 0,12 y un 71% de los pacientes presentó una puntuación estable o mejor después del retratamiento. El perfil de seguridad del retratamiento no se modificó respecto a los primeros 2cursos.
En la actualidad, se pueden considerar hasta 2 cursos adicionales de tratamiento si fuera necesario: tercer o cuarto curso--> (12mg/día en 3días consecutivos) administrados al menos 12 meses después del curso de tratamiento anterior en pacientes con enfermedad activa30.
Situaciones especialesVacunación de convivientesRecomendación 25- •
Los convivientes y personas del entorno más cercano de los pacientes en tratamiento con inmunosupresores como alemtuzumab pueden ser una fuente potencial de contagio. En pacientes que tengan una serología negativa y ya hayan iniciado tratamiento con alemtuzumab, se aconseja evaluar la vacunación frente a varicela o sarampión en los convivientes susceptibles.
- •
Hay datos limitados relativos al uso de alemtuzumab en mujeres embarazadas. Solo debe administrarse alemtuzumab durante el embarazo si los posibles beneficios justifican los riesgos potenciales para el feto30.
- •
Las mujeres en edad fértil en las que se administre alemtuzumab deben tomar medidas contraceptivas adecuadas durante los 4 meses siguientes a la perfusión30.
- •
Se desconoce si alemtuzumab se excreta a la leche materna. No se puede excluir el riesgo para niños lactantes. Por tanto, la lactancia materna debe interrumpirse durante cada curso de tratamiento con alemtuzumab y durante los 4 meses después de la última perfusión de cada uno30.
No se ha establecido todavía la seguridad y eficacia de alemtuzumab en niños con EM de 0 a 18 años de edad. No existe una recomendación de uso específica para alemtuzumab en niños entre 0 y 10 años30.
Intervenciones quirúrgicasRecomendación 29- •
Dado el estado de inmunosupresión transitoria que produce alemtuzumab, parece razonable demorar cualquier tipo de intervención quirúrgica, si la situación clínica motivo de la cirugía lo permite, hasta una reconstitución inmunológica sistémica adecuada. Si el paciente necesita una intervención quirúrgica urgente durante el periodo de linfopenia severa, se aplicará el protocolo local que habitualmente se adopte en pacientes inmunosuprimidos.
- •
La elevada eficacia de alemtuzumab y su rápida implantación en el tratamiento de la EM requiere de un consenso en cuanto al manejo clínico práctico de los pacientes que reciben este tratamiento.
- •
El presente consenso sobre la utilización de alemtuzumab en EM no supone una normativa para su manejo: se ha elaborado con el objetivo de optimizar y facilitar su uso y seguimiento en la práctica clínica habitual.
- •
Un panel de expertos en EM ha consensuado un total de 29 recomendaciones con relación al tratamiento con alemtuzumab, entre las que destacan las siguientes: características de los pacientes candidatos, medidas de seguridad, vacunaciones en pacientes y convivientes, riesgo de infecciones, tratamientos previos y concomitantes y periodos de lavado, medidas contraceptivas, pruebas complementarias (electrocardiograma y radiografía de tórax), citologías con determinación del virus del papiloma humano, RAP, embarazo y lactancia y medidas de contracepción, intervenciones quirúrgicas, perfusión del fármaco y administración de cursos sucesivos al primer curso de tratamiento.
- •
El presente documento de consenso no contempla el manejo de los posibles efectos adversos. En este sentido, otros documentos de consenso sobre el manejo de los efectos adversos autoinmunes con alemtuzumab abordan este tema44-47.
La elaboración de este manuscrito ha sido financiada por Sanofi-Genzyme.
Conflicto de interesesJosé E. Meca-Lallana ha recibido honorarios como consultor o ponente de Biogen-Idec, Celgene, Sanofi-Genzyme, Merck-Serono, Novartis, Roche y Teva; María Fernández-Prada ha participado en actividades docentes apoyadas por Pfizer, ha participado en paneles de expertos promovidos por Sanofi-Genzyme y GSK y ha colaborado en la elaboración de material didáctico con GSK; Elisa García Vázquez ha recibido honorarios por servicios de consultoría de Sanofi-Genzyme; Santiago Moreno Guillén ha recibido apoyo para la investigación y ha participado como ponente en actividades organizadas por Abbvie, Boehringer&Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Janssen Cilag, Merck Sharp&Dohme, Pfizer, Roche y Viiv Healthcare; Susana Otero Romero ha recibido honorarios por servicios de consultoría de Sanofi-Genzyme y honorarios por ponencias de Biogen-Idec y Merck Sharp&Dohme, así como ayudas para la investigación de Novartis; Macarena Rus Hidalgo ha recibido honorarios por servicios de consultoría de Novartis, Merck-Serono, Sanofi-Genzyme, Biogen-Idec, Almirall y Bayer; Luisa M. Villar Guimerans ha recibido honorarios por ponencias, ayuda a la investigación, asistencia a congresos, reuniones de expertos de Merck, Biogen, Sanofi-Genzyme, Roche y Novartis; Sara Eichau Madueño ha recibido honorarios como asesora y ponente de Biogen-Idec, Novartis, Sanofi-Genzyme, Merck-Serono, Roche y Almirall; Óscar Fernández ha recibido honorarios como consultor y por participar en reuniones como moderador o conferenciante y ha participado en ensayos clínicos y otros proyectos de investigación promovidos por Bayer, Biogen-Idec, Merck-Serono, Teva, Novartis, Allergan, Almirall, Sanofi-Genzyme y Roche; Guillermo Izquierdo Ayuso ha recibido honorarios por servicios de consultoría o ponencias de Bayer, Biogen-Idec, Novartis, Sanofi-Genzyme, Merck-Serono, Almirall, Roche, Actelion, Celgene y Teva. Ha recibido también honorarios para proyectos de investigación de Bayer, Biogen-Idec, Novartis, Sanofi-Genzyme, Merck-Serono, Almirall y Teva; José Carlos Álvarez Cermeño ha recibido honorarios por servicios de consultoría de Sanofi-Genzyme, Celgene y Merk; Carmen Arnal García ha recibido honorarios por servicios de consultoría de Sanofi-Genzyme; Rafael Arroyo González ha sido ponente en reuniones con Sanofi-Genzyme, Novartis, Teva, Roche, Biogen-Idec, Merck-Serono y Almirall; Luis Brieva Ruiz ha recibido financiación para proyectos de investigación de su grupo o en forma de honorarios por conferencias, tutorías y ayuda para asistencia a congresos de Bayer, Biogen-Idec, Roche, Merk, Novartis, Allmirall y Sanofi-Genzyme; Carmen Calles Hernández ha recibido honorarios por servicios de consultoría de Sanofi-Genzyme; Antonio García Merino ha recibido honorarios por gastos de viajes y ponencias y asesoría de Bayer, Merck-Serono, Teva, Biogen-Idec, Novartis, Roche, Almirall, Sanofi-Genzyme, así como ayudas para la investigación de Novartis y Biogen-Idec; Montserrat González Plata ha recibido honorarios por ponencias y viajes a congresos de Sanofi-Genzyme, Novartis, Roche y Merck; Miguel Ángel Hernández Pérez ha recibido honorarios por servicios de consultoría de Almirall, Biogen, Merck, Novartis, Roche, Teva y Sanofi-Genzyme; Ester Moral Torres ha recibido honorarios como consultora, asesora y como conferenciante y ha participado en ensayos clínicos y otros proyectos de investigación promovidos por Bayer, Biogen-Idec, Merck-Serono, Teva, Novartis, Almirall, Sanofi-Genzyme, Actelion y Roche; Javier Olascoaga Urtaza ha recibido honorarios por servicios de consultoría de Biogen-Idec, Novartis, Roche y Sanofi-Genzyme y por su participación en reuniones, como organizador, conferenciante o moderador y por ensayos clínicos o proyectos de investigación de Almirall, Bayer, Biogen-Idec, Merck-Serono, Novartis, Roche, Sanofi-Genzyme y Teva; Pedro Oliva Nacarino ha recibido honorarios por servicios de consultoría de Biogen-Idec, Bayer y Sanofi-Genzyme, así como por charlas y conferencias de Biogen, Novartis, Roche, Merck-Serono, Sanofi-Genzyme, Teva y Almirall; Celia Oreja-Guevara ha recibido honorarios por asesorías y conferencias de Biogen-Idec, Merck-Serono, Novartis, Roche, Sanofi-Genzyme y Teva; Rafa Ortiz Castillo es asesor médico de Sanofi-Genzyme; Agustín Oterino ha recibido honorarios como conferenciante de Sanofi-Genzyme, Merck-Serono, Biogen-Idec, Almirall, Allergan y Teva y ha recibido una beca de investigación a través del IDIVAL por Novartis y Sanofi-Genzyme; José María Prieto González es consultor de Bayer, Biogen-Idec, Sanofi-Genzyme, Novartis, Sanofi-Aventis, Teva, Roche, Merck-Serono y Almirall. Ha intervenido como conferenciante en reuniones o simposios organizados por Almirall, Bayer, Biogen-Idec, Sanofi-Genzyme, Merck-Serono, Novartis, Sanofi-Aventis y Teva. Ha recibido financiación de Almirall, Biogen-Idec, Novartis y Sanofi-Genzyme para la realización de proyectos de investigación; Lluís Ramió-Torrentá ha recibido honorarios por servicios de consultoría, ponencias y gastos de viaje por participar en reuniones científicas de Biogen, Merck, Teva, Sanofi-Genzyme, Roche, Bayer, Almirall y Mylan; Alfredo Rodríguez-Antigüedad ha recibido honorarios como ponente o moderador en reuniones científicas, o por su participación en ensayos clínicos y otros proyectos de investigación promovidos por Bayer-Schering, Biogen-Idec, Sanofi-Genzyme, Merck, Novartis, Teva y Roche; Albert Sáiz ha recibido honorarios por ponencias y trabajos de consultoría de Bayer-Schering, Merck-Serono, Novartis, Teva, Sanofi-Genzyme, Biogen-Idec y Roche; Mar Tintoré ha recibido honorarios por servicios de consultoría y ponencias de Almirall, Bayer, Schering, Biogen-Idec, Sanofi-Genzyme, Merck-Serono, Novartis, Roche, Sanofi-Aventis y Teva; Xavier Montalbán Gairin ha recibido honorarios por participar en reuniones científicas y ha formado parte del comité organizador y asesor de ensayos clínicos de Actelion, Bayer, Biogen-Idec, Celgene, Merck-Serono, Novartis, Roche, Sanofi-Genzyme, Teva, Excemed, MSIF y NMSS.
Los autores agradecen a Mónica Giménez su ayuda en los procedimientos de redacción, revisión y envío del manuscrito.

