



Mitochondrial DNA deletion syndromes constitute rare processes resulting from defects in oxidative phosphorylation.1 The incidence of these processes is unknown, but is estimated at up to one case per 5000 live births, with the majority being sporadic processes.2 This syndrome includes 3 overlapping phenotypes: Pearson syndrome, progressive external ophthalmoplegia, and Kearns-Sayre syndrome.1,3–6
Kearns-Sayre syndrome is characterised by the development of progressive external ophthalmoplegia, ptosis, and retinitis pigmentosa with onset before the age of 20.4,6 It is usually associated with such other neurological manifestations as cerebellar ataxia and sensorineural hearing loss. Up to 50% of patients present heart disorders, with the most frequent being conduction disorders. Patients may also display such endocrine manifestations as short stature, hypoparathyroidism, gonadal failure, and diabetes mellitus.7–9
Diagnosis is clinical, and based on the identification of the classic clinical characteristics together with one or more of the following factors: high cerebrospinal fluid (CSF) protein levels (> 100 mg/dL), atrioventricular block, or cerebellar ataxia.5 Muscle biopsy and genetic studies help confirm the diagnosis.5
There is currently no specific treatment for this entity, but clinical improvement has been described after the administration of folinic acid to patients with Kearns-Sayre syndrome,4 since this condition has been associated with cerebral folate deficiency.4,5
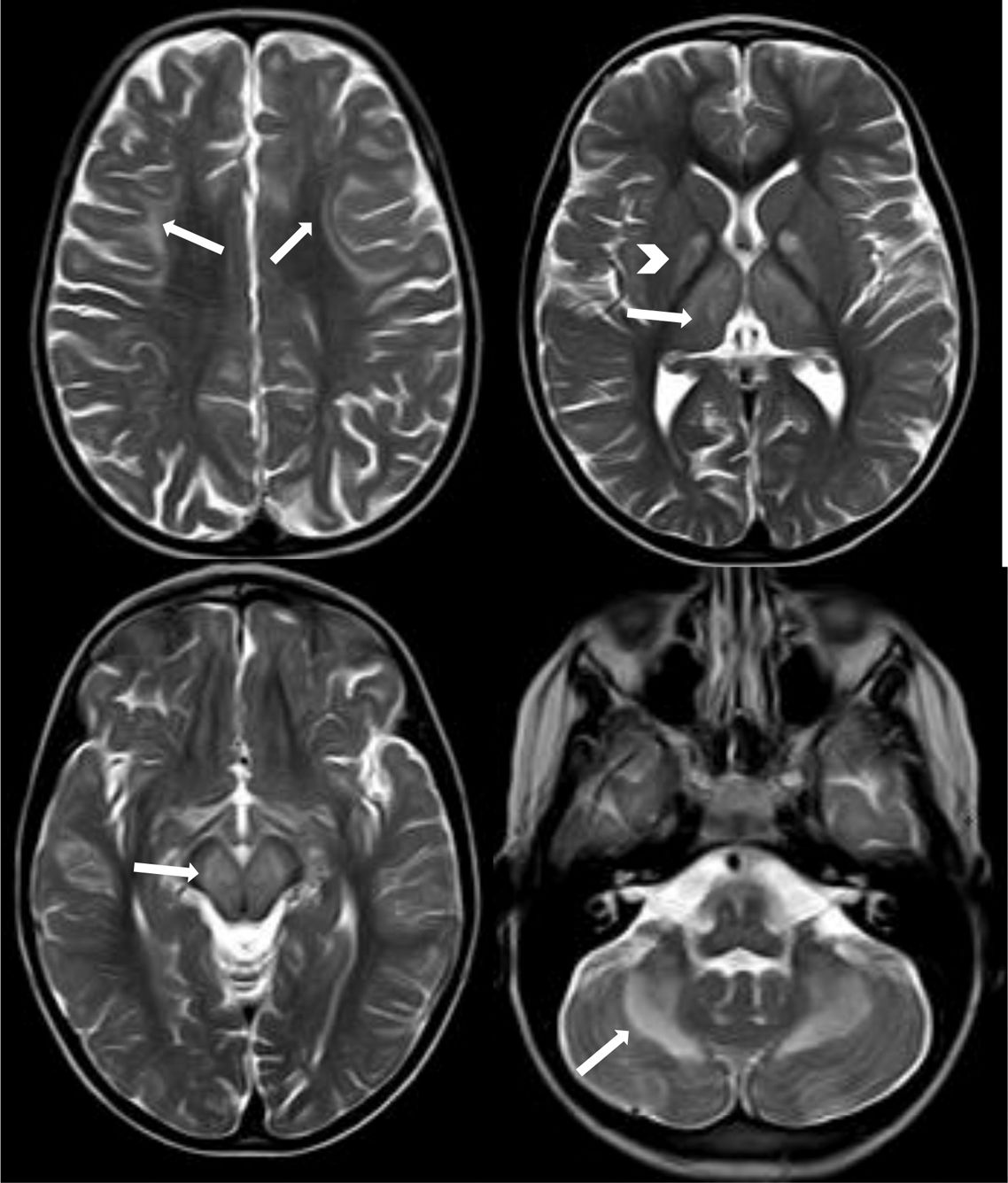
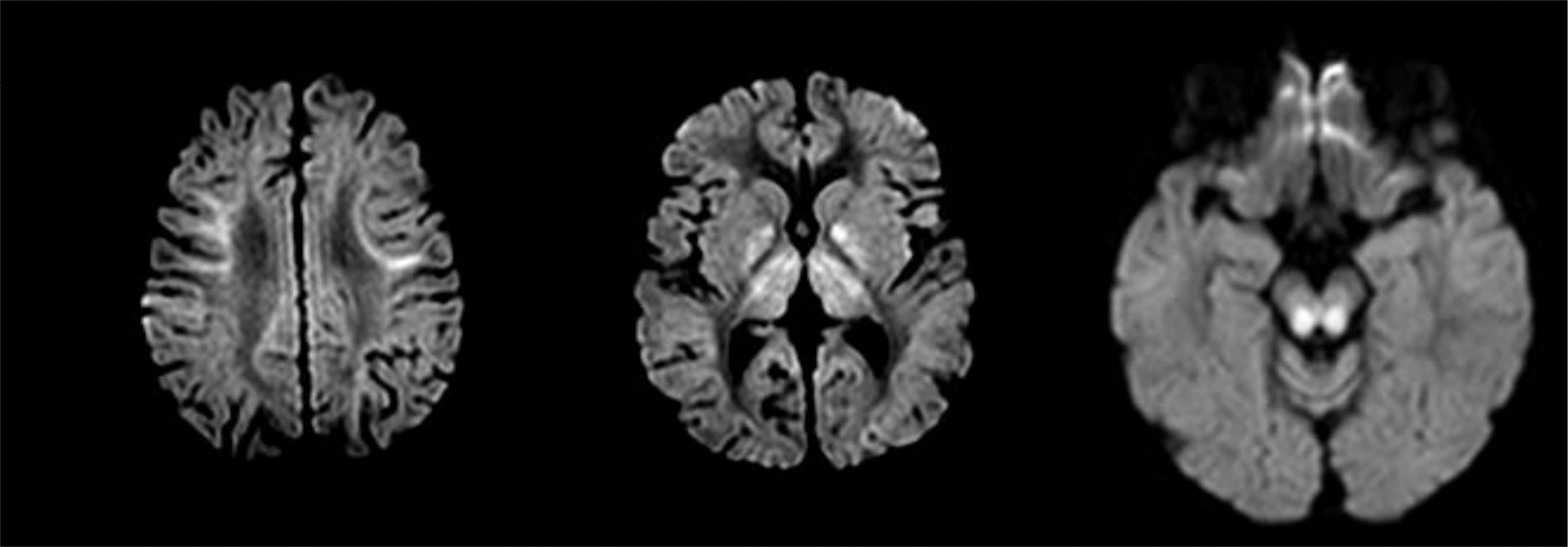
We present the case of an 8-year-old boy displaying difficulties in gross motor skills and intentional tremor, with onset several months earlier. He had been assessed by the paediatric neurology department at the age of 2 due to initially unilateral ptosis which progressed bilaterally. Neuromuscular disorder was ruled out on several occasions (creatine kinase and anti-acetylcholine receptor antibody determinations, negative results for anti-MuSK antibodies, and negative Tensilon test). The patient's personal history also included thrombocytopaenia secondary to bone marrow hypoplasia, hypoparathyroidism, short stature, and primary adrenal insufficiency. Physical examination revealed bilateral ptosis predominating on the right side, limited facial mimicry, proximal muscle weakness, abolished deep tendon reflexes, and action tremor in both hands, hindering his ability to write. We requested a brain magnetic resonance imaging study, which revealed mild cerebral and cerebellar cortical atrophy with bilateral, symmetrical involvement of subcortical fibres of the supratentorial region; the globi pallidi; the thalami; the cerebellar white matter; and very extensive brainstem atrophy (Fig. 1). Areas of increased diffusion were observed in the cerebellar lesion, with restriction in the other areas, possibly representing more acute involvement (Fig. 2). Given the radiological findings supporting the clinical suspicion of Kearns-Sayre syndrome, we requested a blood mitochondrial DNA study, which detected a single 6.4-Kb deletion and a heteroplasmy rate of 27%, confirming the diagnosis. Laboratory testing revealed a low CSF L-5-methyltetrahydrofolate (5-MTHF) level (1.4nmol/L; normal range: 47-90); we therefore started treatment with oral folinic acid. Although CSF 5-MTHF levels normalised at one year of treatment, the patient presented no clinical or radiological improvement; unlike the cases reported in the literature, we have observed progressive deterioration of his baseline situation, with muscle weakness and cerebellar ataxia increasing in severity.
, which is more pronounced in the frontal lobes (arrows in the upper left image), globi pallidi (arrowhead in the upper right image) and thalami (arrow in the upper right image), brainstem (arrow in the lower left image), and cerebellar white matter, with bilateral, symmetrical involvement.")
Axial T2-weighted TSE sequence showing signal hyperintensity in the subcortical white matter (U-shaped fibres), which is more pronounced in the frontal lobes (arrows in the upper left image), globi pallidi (arrowhead in the upper right image) and thalami (arrow in the upper right image), brainstem (arrow in the lower left image), and cerebellar white matter, with bilateral, symmetrical involvement.
 showing diffusion restriction in the affected areas.")
During follow-up, he developed bilateral sensorineural hearing loss; complete atrioventricular block, which required an emergency pacemaker implantation; retinitis pigmentosa; and progressive cerebellar syndrome with gait loss.
Please cite this article as: Pardo Ruiz E, Maturana Martínez D, Vázquez López M, Ruiz Martín Y. Síndrome de Kearns-Sayre: ausencia de respuesta clínica al tratamiento con ácido folínico oral. Neurología. 2019;34:618–620.
