



Stroke is one of the leading causes of death in the world; its incidence is increasing due to increased life expectancy. However, treatment options for these patients are limited since no clinically effective drugs have been developed to date.
DevelopmentAccording to clinical evidence, a number of neurochemical changes take place after stroke, including energy depletion, increased free radical synthesis, calcium accumulation, neurotransmitter imbalance, excitotoxicity, and, at a later stage, immune system activation leading to inflammation.
Immune response has been shown to be a major factor in disease progression. The release of proinflammatory cytokines such as TNF increase brain damage secondary to excitotoxicity and calcium accumulation, and promote free radical synthesis and cell death through various mechanisms. On the other hand, certain anti-inflammatory cytokines, such as IL-10 and IL-4, have been shown to have a neuroprotective effect and even promote neurogenesis and synapse remodelling, which makes immune modulation a promising treatment approach.
ConclusionsUnderstanding the relationship between the immune system and the nervous system not only deepens our knowledge of stroke but also provides new diagnostic, prognostic, and therapeutic strategies that may increase the quality of life of stroke patients.
El ictus es una de las principales causas de mortalidad en el mundo y debido al incremento en la expectativa de vida su incidencia va en aumento; sin embargo, el desarrollo de nuevos medicamentos con utilidad clínica ha sido prácticamente nulo, por lo que hasta la fecha el tratamiento de estos pacientes es muy limitado.
DesarrolloLa evidencia básica y clínica en el área señala que tras un infarto cerebral se producen una serie de cambios neuroquímicos, entre los que se encuentran: la depleción energética, la producción de radicales libres, la acumulación de calcio, la desregulación de neurotransmisores, la excitotoxicidad, y de manera tardía, la activación del sistema inmune caracterizada como inflamación. Esta respuesta del sistema inmunológico ha mostrado ser un evento central en la progresión de la patología, en el que destaca la participación de las citocinas proinflamatorias como TNF, que aumentan el daño por excitotoxicidad y por acumulación de calcio, favorecen la formación de radicales libres y en general promueven la muerte celular. Por otro lado, algunas citocinas antiinflamatorias como IL-10 e IL-4 han mostrado tener efectos neuroprotectores e incluso favorecen la recuperación de sinapsis y la neurogénesis, haciendo de la modulación de la respuesta inmunológica un área con mucho potencial terapéutico.
ConclusionesEl entendimiento de las relaciones entre el sistema inmunológico y el sistema nervioso no solo nos permite entender con mayor profundidad el fenómeno del ictus, sino que también nos ofrece un nuevo arsenal de estrategias diagnósticas, pronósticas y terapéuticas que podrían mejorar la calidad de vida de las personas aquejadas por esta terrible enfermedad.
Stroke is the second leading cause of death and the third most frequent cause of disability worldwide. Ischaemic stroke is estimated to affect 16 million people each year, of whom 6 million die; most stroke survivors are left with severe sequelae. The incidence of stroke is increasing, particularly in developing countries.1 Ischaemic stroke is characterised by the interruption of blood supply to a brain region, most frequently due to a clot or thrombus obstructing a cerebral blood vessel. This results in neuron death in that brain area, known as the ischaemic core; the surrounding, hypoperfused tissue is known as the ischaemic penumbra. The ability of the ischaemic penumbra to recover from stroke largely determines the patient's prognosis and functional recovery.2
In this article, we review the association between immune system activation and the outcome of the ischaemic penumbra, and describe immune system alterations in response to stroke.
Pathophysiology of ischaemic strokeThis section briefly outlines the pathophysiological mechanisms of ischaemic stroke; for further insight into the topic, we recommend references 3, 5, 6, 7, and 8.
Interruption of blood supply results in a lack of oxygen and glucose in the brain tissue, leading to fast depletion of adenosine triphosphate (ATP), the body's main medium for energy exchange.3 ATP depletion prevents the cell from performing basic functions: sodium-potassium pump (Na+/K+-ATPase) activity is suppressed, leading to resting potential alterations and intracellular Na+ accumulation. This, in turn, results in anoxic depolarisation and cytotoxic oedema.4 Anoxic depolarisation spreads across the penumbra,4 leading to the uncontrolled release of such neurotransmitters as glutamate,5 which is thought to increase the damage to brain tissue.4
Depolarisation also significantly increases glutamate concentration. This neurotransmitter activates the NMDA receptor, increasing the conductivity of Na+; the ion's osmotic effects result in cytotoxic oedema. NMDA receptor hyperactivity increases the concentration of Ca2+, which may cause mitochondrial dysfunction due to increased permeability, increased free radical production, and activation of phospholipases and caspases, leading to cell death by apoptosis.5,6
The cellular components of damaged cells may subsequently activate immune cells, promoting cytokine release and the synthesis of such free radicals as nitric oxide (NO), which is eventually metabolised to nitrite (NO2) and hydroxyl radical (•OH).7 This, together with the damage caused by ischaemia, may result in loss of selective permeability of the blood-brain barrier (BBB), allowing potentially toxic substances to enter the central nervous system, and recruiting a greater number of immune system cells through adhesion molecules (ICAM and VCAM).8,9
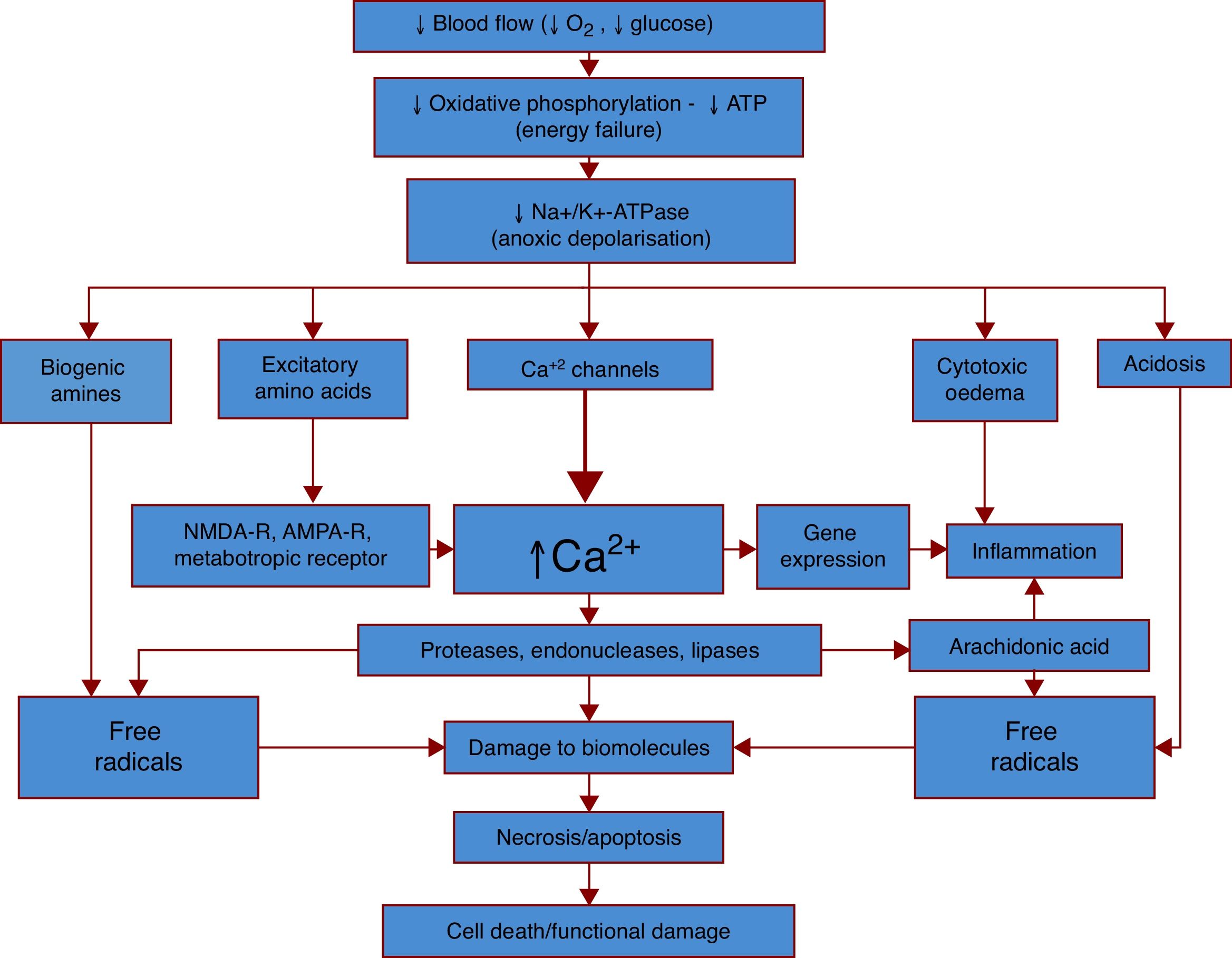
Fig. 1 provides a simplified explanation of the pathophysiology of ischaemic stroke.
 production, leading to dysfunction of numerous processes. Dysfunction of the sodium-potassium pump (Na+/K+-ATPase), one of the main processes, results in anoxic depolarisation. Uncontrolled depolarisation leads to the accumulation of neurotransmitters and other amines in neurons, which in turn increases levels of free radicals, calcium, water, and hydrogen ions. This triggers a strong inflammatory response, which results in cell death and functional damage.")
The general pathophysiological mechanisms of ischaemic stroke. An ischaemic event begins with a considerable decrease in cerebral blood flow, which reduces the supply of oxygen and glucose, the main sources of energy for neurons. Oxygen/glucose depletion results in reduced energy (ATP) production, leading to dysfunction of numerous processes. Dysfunction of the sodium-potassium pump (Na+/K+-ATPase), one of the main processes, results in anoxic depolarisation. Uncontrolled depolarisation leads to the accumulation of neurotransmitters and other amines in neurons, which in turn increases levels of free radicals, calcium, water, and hydrogen ions. This triggers a strong inflammatory response, which results in cell death and functional damage.
Cell damage and the release of some cellular components trigger an inflammatory response at the site of the lesion; the components of this reaction play a major role in the modulation of the inflammatory response in the area of penumbra, either aggravating the damage or exerting a neuroprotective effect.
One of the first stages of inflammatory response is the detection of damage-associated molecular patterns (DAMP) by toll-like receptors (TLR) of the innate immune system, present mainly in microglia but also in endothelial cells and astrocytes.10,11 The activation of several of these receptors leads to the secretion of important components of the immune response, including cytokines, chemokines, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2), which in turn increases NO, prostaglandin E2 (PGE2), and myeloperoxidase levels and results in the recruitment of other cellular and non-cellular components of the immune system.10,12
The importance of TLRs has been shown in experimental studies of middle cerebral artery obstruction: TLR4-deficient mice display smaller infarctions and better neurological status according to a standardised test battery; reduced iNOS, COX-2, and gelatinase-B (a protease involved in haemorrhagic transformation of stroke) activity; and lower levels of malondialdehyde (an important marker of free radical activity). No significant differences in cytokine levels were found between mutant and control mice, however.12
TLR4 deficiency has a protective effect in mutant mice. However, when lipopolysaccharide (a component of bacteria that activates TLR4; LPS) is administered before ischaemia, TLR4 has a neuroprotective effect; this phenomenon is known as LPS preconditioning.13
During preconditioning, exposure to LPS modifies TLR4 response by recruiting the TRIF/IRF3 signalling pathway rather than the MyD88, the normal signalling pathway for this receptor. This increases the concentration of interferon beta (IFN-β), which has a powerful neuroprotective effect.13
The following sections address specific components of immune response and their role in stroke.
AstrocytesAs an integral part of synapses, astrocytes respond early to the ischaemic event. As mentioned previously, these cells may be activated by molecules associated with cell damage via TLRs and by the presence of cytokines in the medium. In both mechanisms, the ischaemic event induces astrogliosis in astrocytes; second messenger STAT3 plays an essential role as it causes a phenotypic change.14
Astrogliosis can isolate the damaged tissue, preventing propagation of the damage. A band is created around the site of infarction that captures proinflammatory cells, prevents growth of the infarcted area, and improves functional prognosis in animals.14,15
STAT3 ablation associated with glial fibrillary acidic protein (GFAP), the glia-specific second messenger, reduces astrocyte migration and these cells’ ability to isolate damaged tissue in a model of spinal cord injury.14 A mouse model of Socs3 (the main STAT3 inhibitor) inhibition increased astrocyte activity, and mice displayed enhanced contraction of the damaged tissue and improved functional recovery.16
Astrocytes mediate a wide range of neuroprotective mechanisms. These include activation of such antioxidants as glutathione17; secretion of such neurotrophins as GDNF, which inhibit apoptosis18,19; and the metabolism of numerous neurotransmitters, mainly glutamate, preventing excitotoxicity.20,21 A detailed review of the function of astrocytes and the changes they display is provided in reference 20.
The latter mechanism has been exploited to great effect: GLT1 overexpression reduces the volume of the infarcted area in a rat model of stroke, improving functional prognosis by reducing glutamate release to the healthy tissue and cell death after middle cerebral artery occlusion.21
Several drugs share this action mechanism, constituting a promising area of research: tamoxifen,22 used as a prophylactic treatment for breast cancer; riluzole,23 used in amyotrophic lateral sclerosis; and ceftriaxone.24 This antibiotic has been shown to improve survival and functional recovery in animal models of stroke.24 Despite its protective role, astroglia can also increase damage due to secretion of proinflammatory cytokines, such as tumour necrosis factor (TNF) or IL-6, and chondroitin sulphate proteoglycans, which inhibit axonal growth and synaptogenesis. Administration of chondroitinase has been shown to improve functional recovery after spinal cord injury.25
Microglia and macrophagesMicroglial cells constitute the main component of the immune system in the brain. Under physiological conditions, microglia move extensively throughout the brain and respond progressively to different signals of injury.26 Some of the main factors determining microglial function include the microenvironment: stimulation with IFN-γ causes microglial polarisation to the highly proinflammatory M1 phenotype, which is associated with neurodegeneration, whereas IL-4 induces microglial differentiation into M2, a phenotype that promotes neurogenesis and oligodendrogenesis and has anti-inflammatory properties.26,27
In spite of these opposing roles, microglial cells seem to have a protective effect in stroke. In a study of transgenic mice expressing a mutant thymidine kinase form of herpes simplex virus driven by myeloid-specific CD11b promoter, Lalancette-Hébert et al.28 used ganciclovir to ablate proliferating microglia, observing an increase in the size of the ischaemic lesion, the number of apoptotic neurons, and in behavioural alterations; this was not observed in control mice receiving ganciclovir.
A mouse model of middle cerebral artery occlusion revealed a prevalence of M2-type genes from day 1 to day 5 after ischaemia, whereas expression of M1-type genes began on day 3 and continued until day 14.27 This is especially significant considering the results of in vitro studies of neurons subjected to oxygen-glucose deprivation, which show that neuronal survival decreases considerably in the presence of M1-polarised microglia, whereas M2-polarised microglia increase survival.27
From a treatment viewpoint, the antibiotic minocycline has achieved the most significant advances in microglial cells; it has been found to have neuroprotective effects, to reduce neuronal apoptosis and infarct volume, and to improve functional recovery in mice.29
Given the drug's safety profile and the available clinical experience, minocycline has been studied extensively in humans as a neuroprotective treatment for stroke, with mixed results.30,31
Other cellular componentsInflammation and BBB disruption allow cells from the peripheral immune system to enter the brain, modifying the course of the disease. Neutrophil recruitment is observed as early as 24 hours after stroke.32 Macrophage recruitment occurs at days 3 to 7 after stroke.33 Other components, such as T- and B-cells, are recruited at later stages. All these cell types have been observed to be involved in the pathophysiology of stroke.
Neutrophils have been shown to be essentially neurotoxic. In humans, high plasma neutrophil counts have been correlated with poorer prognosis following stroke.34 In animal models, treatment with anti-neutrophil monoclonal antibody (RP3) prevents neutrophil infiltration to the central nervous system, reduces oedema, and decreases infarct volume.35,36
Neutrophil activation results in permanent BBB disruption; this may in part be explained by the release of gelatinase (matrix metalloproteinase 9), which is generated mainly by neutrophils.36,37 The presence of gelatinase and BBB disruption have been shown to play an essential role in the haemorrhagic transformation of ischaemic stroke both in animals and in humans.38,39
Mutant mice overexpressing tissue inhibitor of matrix metalloproteinase 1 and undergoing carotid artery occlusion display smaller infarcts, greater BBB integrity, and reduced leucocyte infiltration compared to wild-type mice.39,40
Unlike other immune cells, lymphocytes have been associated with better recovery after stroke, according to NIHSS scores.34 T helper cells are the most frequently studied type of lymphocyte. As occurs with microglia, polarisation of T helper cells determines their response: Th1 cells secrete mainly TNF, IFN, and IL-6, whereas Th2 cells secrete IL-4.41
This functional differentiation not only affects stroke progression and functional recovery (with Th2 being neuroprotective), but also directs the peripheral immune response to other sites of stroke. Following stroke, there is a decrease in the number of T cells in the spleen, thymus, lymph nodes, and intestinal barrier, possibly due to a high level of apoptosis resulting from sympathetic nervous system activity. Th2 polarisation and the reduction in the number of lymphocytes favours infection; this is known as stroke-induced immunodeficiency.42–44
Tumour necrosis factor alphaTNF is one of the first cytokines to be released during stroke; this has important implications for cell survival or death.
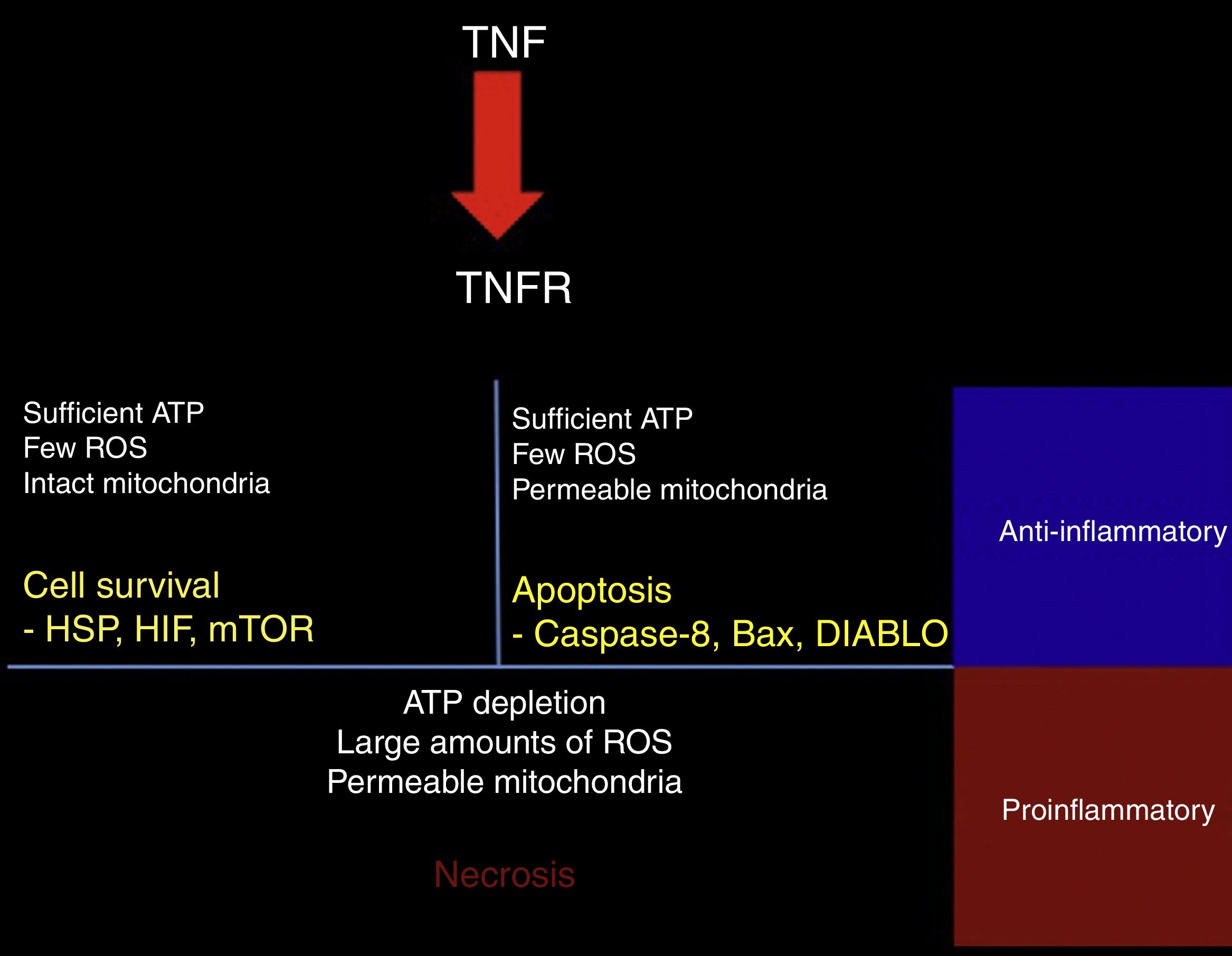
TNF-α may have protective or cytotoxic effects depending on the cell medium. A mathematical model has shown that activation of the TNF receptor leads to the activation of one of 3 signalling pathways. The first pathway, RIP1-NFkB, is associated with cell survival, inhibition of apoptosis and necrosis, and increased resistance to subsequent damage. This pathway is promoted by a medium with low reactive oxygen species (ROS) activity, high ATP levels, and high NFkB activity (Fig. 2).45

Response to TNF. TNF activates different intracellular signalling mechanisms according to the cell's metabolic state. If the cell still has sufficient ATP and few reactive oxygen species (ROS), and the mitochondrial membrane remains impermeable, the cell activates heat shock proteins (HSP), hypoxia-inducible factor (HIF), and the mammalian target of rapamycin (mTOR). When the cell has sufficient ATP and few ROS, but the mitochondrial membrane is permeable, the proapoptotic pathways are activated via caspase-8, Bax, and DIABLO. ATP depletion, large amounts of ROS, and dysfunctional mitochondria lead to necrosis. In tissues, surviving or apoptotic cells exert an anti-inflammatory and cytoprotective effect, whereas necrosis constitutes a powerful proinflammatory stimulus.
The second pathway is promoted by high ATP concentrations associated with increased mitochondrial permeability and an intermediate concentration of ROS; caspase-8 and caspase-3, the mediators of this pathway, induce apoptosis.45
The third pathway is promoted by high ROS levels, low ATP levels, and increased mitochondrial permeability, leading to increased activity of the RIP3 mediator, which induces necrosis (Fig. 2).45 Cell fate is important for the tissue environment, as cell survival and apoptosis generate anti-inflammatory responses whereas necrosis promotes inflammation.46
In addition to its direct effects on cell survival, TNF-α promotes excitotoxicity47,48; increases calcium influx through expression of the AMPA receptor GluR2 subunit49; promotes the release of TNF, BDNF, and glutamate from microglial cells50,51; and, at certain concentrations, blocks the expression of glutamate transporter GLT1 in astrocytes. This process is reversed by blocking this cytokine or BDNF.52,53
Interferon gammaIFN-γ plays an essential role in the brain: it polarises immune cells towards the M1 phenotype, increases the expression of Fas and TNF receptors (increasing cells’ sensitivity to apoptosis), and increases the activity of free radical–producing enzymes such as iNOS and the synthesis and release of chemokines.54
The chemokines that are overexpressed include IP-10 and CXCL10. The latter attracts Th1 lymphocytes and prevents the activation of Th2 cells.55 CXCL10 also increases IFN-γ production by the microglia, generating positive feedback.55
In animal models, splenectomy or treatment with anti–IFN-γ polyclonal antibodies significantly reduced the size of the infarcted area and improved functional recovery. IL-10 is a promising treatment as it inhibits endogenous production of IFN-γ.54,55
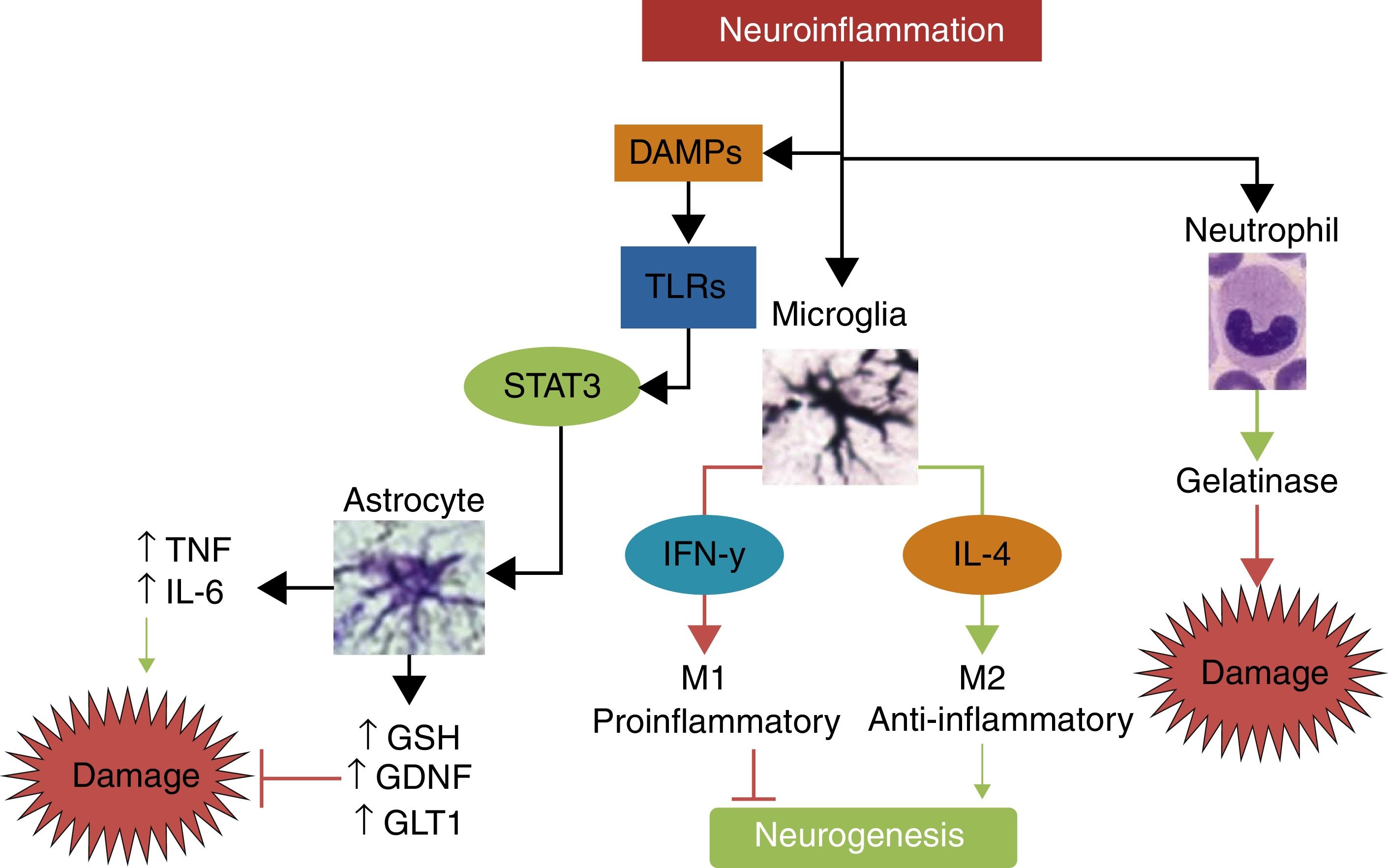
Interleukin-10IL-10 is the main anti-inflammatory cytokine. Its potentially protective effects include polarisation of immune cells towards the M2 phenotype, reducing the migration of proinflammatory cells through the endothelium; promoting the release of other neuroprotective cytokines such as IL-4; decreasing secretion of TNF, IL-1, and IFN; and reducing the production of free radicals and chemokines after stroke.56 In animal models of stroke, IL-10 has been shown to significantly reduce infarct volume and neurological deficits.57,58Fig. 3 summarises the pathways of damage and neuroprotection against neuroinflammation.

The main mechanisms of damage and neuroprotection associated with neurinflammation.Neuroinflammation is not a neurotoxic stimulus in itself; immune response mainly activates the cells of the innate immune system, such as neutrophils, microglia, and astrocytes. Neutrophils are predominantly neurotoxic, whereas microglia and astroglia can express neuroprotective phenotypes. Depending on the type and intensity of the stimulus, microglia may polarise to the M1 phenotype, which releases IFN-γ, leading to greater damage and inhibition of neurogenesis, or to the M2 phenotype, which secretes IL-4 and has anti-inflammatory properties, promoting neurogenesis and neuroprotection. When activated by damage molecules, and depending on which TLR receptor is activated, astroglia may secrete TNF and IL-6, promoting neuronal damage, or such neuroprotective factors as antioxidant GSH, the GDNF growth factor, and the GLT1 transporter.
Increased neurogenesis has been observed in the dentate gyrus during ischaemia, especially in models of focal ischaemia.59–63 However, we are yet to determine whether this activation plays a role in functional recovery after ischaemic stroke. The proliferation and maturation of new neurons depends on several factors, including inflammation.64,65
Studies have shown that the neurogenic response can be modulated by microglial activation during an ischaemic event, but it depends on whether microglia are in an M1 proinflammatory state, which inhibits neurogenesis, or an M2 anti-inflammatory state, which promotes it.66–69
In vivo and in vitro studies have shown that microglial cells at stages 2 and 3 of activation promote neurogenesis in the dentate gyrus by secreting TGF-β70 and IL-4.71 IL-15 plays an important role in the subventricular zone, promoting progenitor cell proliferation and neuronal differentiation.72 IL-15 regulation in a mouse model of Alzheimer disease showed increased neurogenesis in vivo and increased progenitor cell proliferation in vitro.73 By contrast, such factors as TNF-α have been found to inhibit the formation of new neurons in the hippocampus.71,74,75 IL-1 secretion has also been found to inhibit neurogenesis,76,77 whereas IL-1 overexpression in mice reduces neuronal differentiation and the survival of new neurons.78,79
It has recently been suggested that antidepressant pretreatment with selective serotonin reuptake inhibitors (SSRI) may improve the alterations caused by ischaemic stroke in humans80 and in animals.81,82 This process is yet to be fully understood, however. Some researchers suggest that such SSRIs as fluoxetine and citalopram inhibit the release of IL-1β, TNF-α, NO, glutamate, and D-serine by activated microglia, promoting survival of cortical neurons in an in vitro model of hypoxia.83 The same effect has been evaluated in an in vitro model of LPS-induced inflammation, where SSRIs also reduced TNF-α and NO release; this suggests that these drugs may have an anti-inflammatory effect.84 Further studies are necessary to evaluate the potential of antidepressants to treat ischaemic stroke.
ConclusionsOur rapidly increasing understanding of the interactions between the brain and the immune system has shed light on potential new treatment strategies for such highly prevalent conditions as ischaemic stroke.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Sotomayor-Sobrino MA, Ochoa-Aguilar A, Méndez-Cuesta LA, Gómez-Acevedo C. Interacciones neuroinmunológicas en el ictus. Neurología. 2019;34:326–335.
