



Niemann-Pick disease type C (NPC; MIM #257220 and MIM #607625) is caused by an inborn error of metabolism and follows an autosomal recessive inheritance pattern. The disease is characterised by an alteration to intracellular cholesterol transport, causing lysosomal and/or late endosomal accumulation of several metabolites.1,2 The causal genes are NPC1 (MIM *607623) and NPC2 (MIM *607625). NPC1 accounts for 95% of published cases, with NPC2 covering 4%.3
Diagnosis is challenging due to the great clinical heterogeneity of the condition and the difficulty of laboratory testing. Symptoms are generally classified into 3 broad groups: visceral, neurological, and psychiatric. The most frequent symptoms are splenomegaly, vertical gaze palsy, and cataplexy.3 A screening tool based on clinical manifestations (the NPC Suspicion Index) has been created to guide diagnosis; the tool assigns a score reflecting the risk of NPC and the suitability of additional studies to clarify the diagnosis.4,5
We present the case of a 26-year-old woman of Berber origin; she had history of intellectual disability and psychotic disorder, for which she was receiving olanzapine 5mg. She was referred to our centre due to abnormal movement and gait alterations of less than one year's progression. The neurological examination revealed dysarthria, hypomimia, vertical gaze palsy, global bradykinesia with isolated distal myoclonus, and mild ataxic gait without arm swing. Brain MRI and DaTSCAN® revealed no relevant abnormalities. The ophthalmic examination revealed cherry-red spots on both maculae. Abdominal ultrasound detected splenomegaly (13.8cm). The patient scored 104 on the NPC suspicion index (64th percentile).
To confirm the diagnosis, we tested plasma chitotriosidase activity6 and concentrations of the chemokine CCL18 (PARC)7 and 7-ketocholesterol (7-CC)8–10 using methods described elsewhere. All 3 biomarkers were above the intra-laboratory reference limit.
Sequencing of NPC1 and NPC2 was performed according to the Sanger method with small modifications to the protocol published by Zech's study group, identifying 2 variants in NPC1: p.Ile1061Thr (previously described by Yamamoto et al.11) and p.Val856Ala (not previously described).
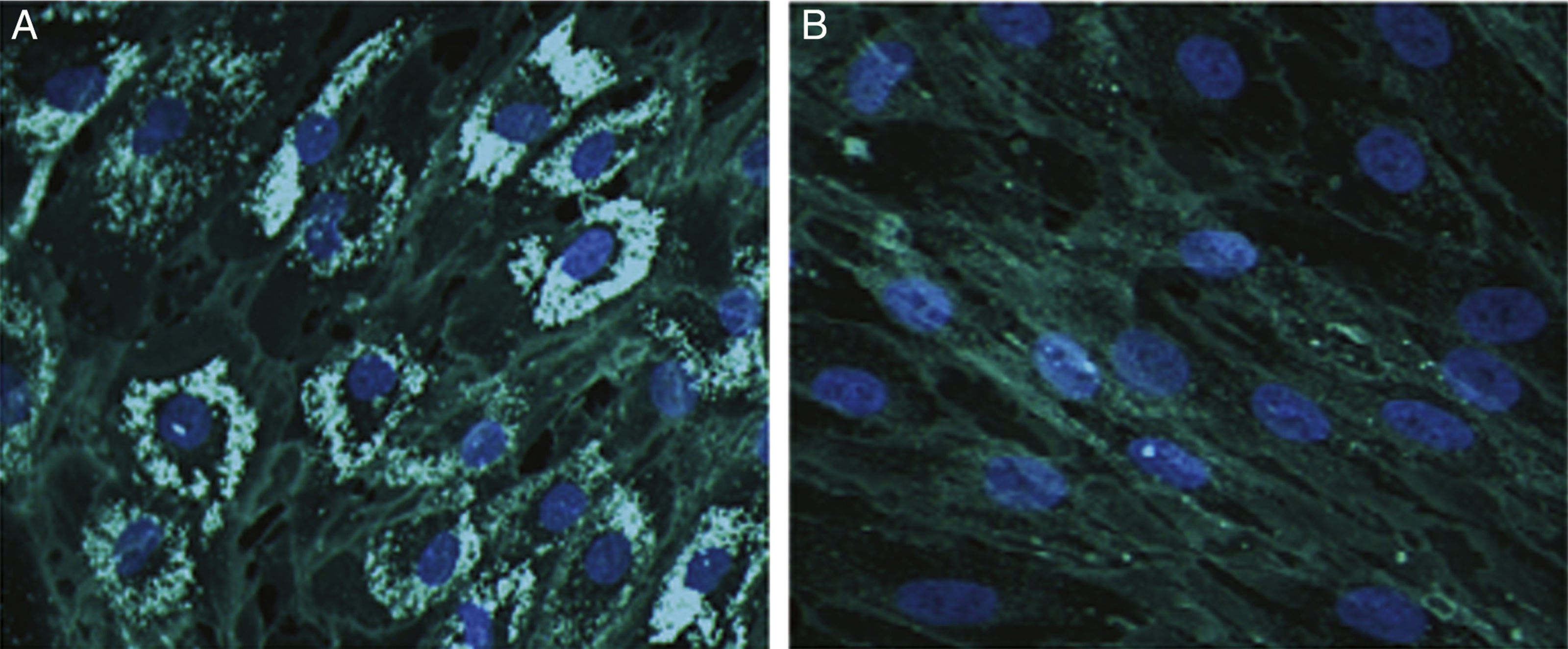
Cholesterol accumulation in fibroblasts was analysed with cytochemical staining using Filipin III (Sigma-Aldrich; Spain)12; 70% of fibroblasts were positive for cholesterol accumulation (Fig. 1).
 and a control (B). Cells were cultured for 72hours on a coverslip in lipoprotein-free medium and then for 24hours in a medium rich in low-density lipoproteins. They were subsequently fixed and stained with Filipin III. 200× magnification.")
Representative images from Filipin III cholesterol staining of fibroblasts from our patient (A) and a control (B). Cells were cultured for 72hours on a coverslip in lipoprotein-free medium and then for 24hours in a medium rich in low-density lipoproteins. They were subsequently fixed and stained with Filipin III. 200× magnification.
To rule out other lysosomal conditions of the same metabolic pathway, we studied lysosomal acid lipase13 and sphingomyelinase14 activity in lysate from leukocytes; results were within the intra-laboratory reference limits.
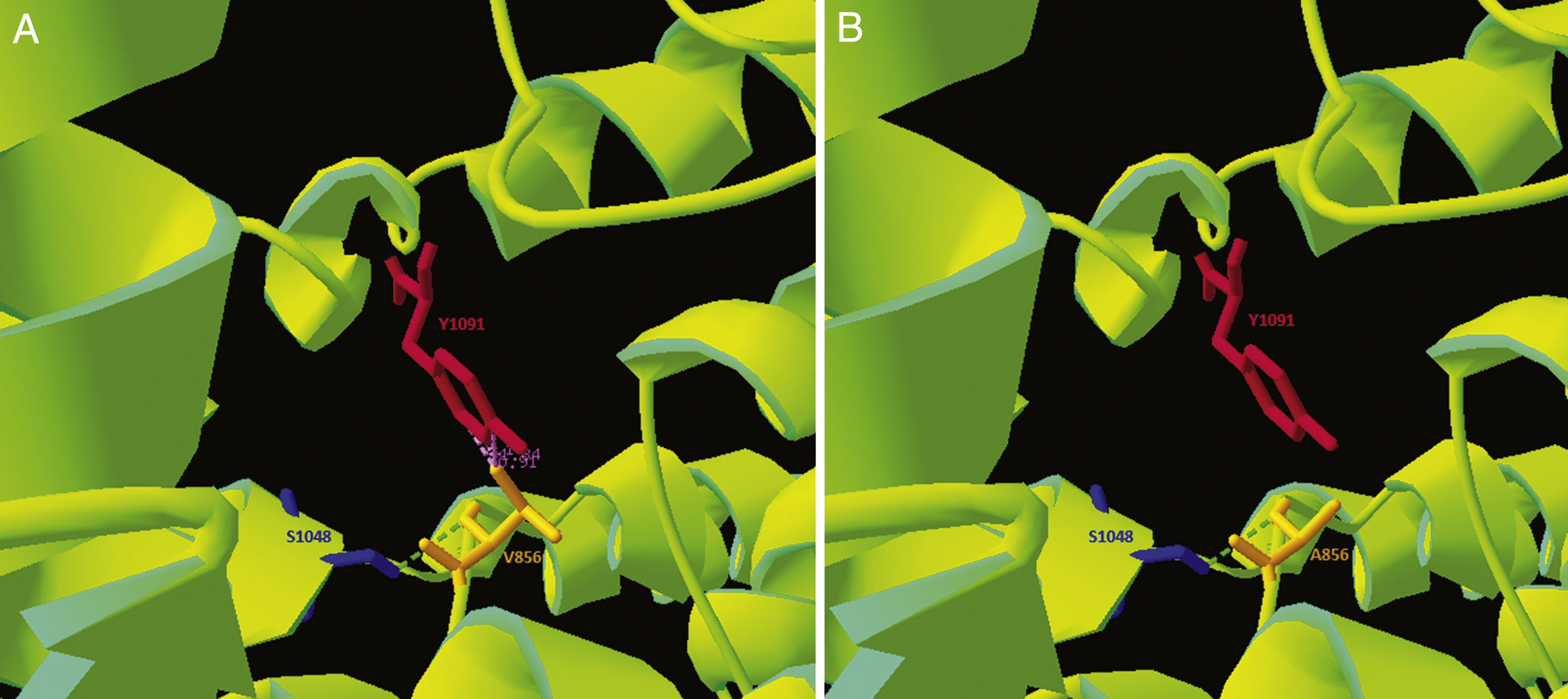
As the p.Val856Ala variant had not previously been described, and given its location in a cysteine-rich loop that accounts for approximately one-third of the described pathogenic variants,1 we performed an in silico analysis of the mutation. Clustal Omega was used to run a sequence conservation analysis, with comparison against 30 species, revealing that the region is highly conserved. Simultaneously, we used the NPC1 structure published on Protein Data Bank by Gong and colleagues in 2016 (Cryo-EM structure of the full-length human NPC1 at 4.4 angstrom; PDB access number: 3JD8) and modelled the pathogenic variant with the free software application Swiss-PdbViewer, revealing reduced electrostatic interactions in the variant form (Fig. 2). The DUET stability predictor detected reduced stability in the protein containing the p.Val856Ala variant (ΔΔG: −1.457 vs ΔΔG: 0kcal/mol). According to the guidelines of the American College of Medical Genetics,15 this variant would be catalogued as “likely pathogenic.”
 or Ala856 (B) amino acids. Images generated using the 3JD8 sequence (PDB) on the Swiss-PdbViewer software.")
The clinical, biochemical, molecular, and cellular findings from our patient suggest that she has NPC due to compound heterozygosis of the p.Ile1061Thr variant and the previously undescribed p.Val856Ala variant.
The authors are grateful to María Teresa Sagrario Fustero, Clementina del Canto Pérez, and Adolfo Mínguez-Castellanos for their help in the clinical diagnosis of the patient, and to Jorge J. Cebolla, Pilar Irún, and Carmen Domínguez for their invaluable contribution in laboratory testing.
Please cite this article as: López de Frutos L, Romero-Imbroda J, Rodríguez-Sureda V, Giraldo P. Nueva variante asociada a enfermedad de Niemann-Pick tipo C: manifestaciones neurológicas y caracterización bioquímica, molecular y celular. Neurología. 2020;35:50–52.
