



La enfermedad de Niemann-Pick tipo C (NPC; MIM#257220 y MIM#607625) se debe a un error innato del metabolismo, de herencia autosómica recesiva, caracterizado por una alteración del tráfico intracelular de colesterol, lo que provoca una acumulación de múltiples metabolitos en el lisosoma y/o endosoma tardío1,2. Los genes responsables son NPC1 (MIM*607623) y NPC2 (MIM*607625). NPC1 engloba el 95% de los casos publicados, mientras que NPC2 es el responsable de un 4%3.
El diagnóstico de la enfermedad de NPC es complejo dado la gran heterogeneidad clínica que presenta y la dificultad de las pruebas de laboratorio. La sintomatología se clasifica en 3 grandes bloques: visceral, neurológica y psiquiátrica, siendo las características más frecuentes la esplenomegalia, la parálisis de la mirada vertical y la cataplejía3. Basándose en las manifestaciones clínicas y con la finalidad de ayudar a orientar el diagnóstico se ha creado una herramienta de cribado que asigna una puntuación en función del riesgo de ser afecto de NPC (NPC índice de sospecha [NPC-SI]), y que sugiere la idoneidad de realizar más estudios para esclarecer el diagnóstico4,5.
Presentamos el caso de una mujer de 26 años de etnia bereber con antecedentes de retraso mental y trastorno psicótico en tratamiento con olanzapina 5mg que es remitida por movimientos anormales y alteración de la marcha desarrollados en el último año. En la exploración neurológica destacaba disartria, hipomimia facial, parálisis de la mirada vertical, bradicinesia global con mioclonias aisladas distales y marcha ligeramente atáxica sin braceo. La RM cerebral y DaTSCAN® no mostraron alteraciones relevantes. El estudio oftalmológico reveló una mancha rojo cereza en la mácula de ambos ojos. Una ecografía abdominal mostró esplenomegalia de 13,8cm. El test de cribado NPC-SI puntuaba 104 (percentil 64).
Para confirmar el diagnóstico de NPC se analizó la actividad plasmática de la quitotriosidasa6 y la concentración de la quimioquina CCL18/PARC7 y 7-cetocolesterol8–10 (7-CC) mediante métodos previamente descritos, obteniendo valores por encima del rango de referencia intralaboratorio (RRI) para los 3 biomarcadores.
La secuenciación de los genes NPC1 y NPC2 se realizó mediante el método de Sanger con pequeñas modificaciones respecto al protocolo publicado previamente por Zech et al., identificando 2 variantes en NPC1: p.Ile1061Thr previamente descrita por Yamamoto et al.11, y p.Val856Ala, no descrita previamente.
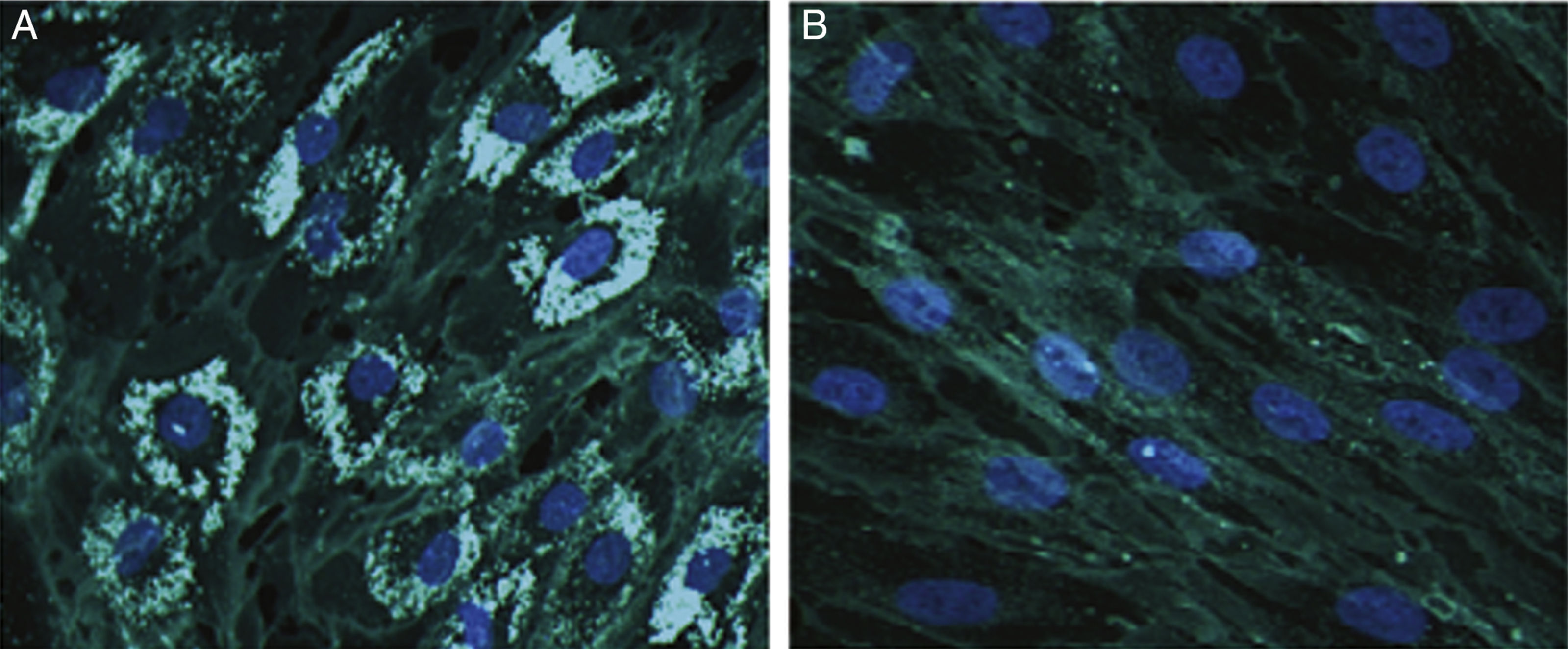
El acúmulo de colesterol en fibroblastos del paciente fue analizado mediante tinción citoquímica por Filipin III12 (Sigma-Aldrich, ES), observándose una positividad del 70% (fig. 1).
 y de un control (B). Las células se cultivas en cubreobjetos y se mantienen durante 72h en un medio libre de lipoproteínas antes de cambiarlas a un medio rico en lipoproteínas de baja densidad durante 24h. Posteriormente se fijan y tiñen con el colorante Filipin III. Ampliación a ×200")
Tinción con Filipin III. Figuras representativas de la tinción del colesterol mediante Filipin III en fibroblastos de nuestro paciente (A) y de un control (B). Las células se cultivas en cubreobjetos y se mantienen durante 72h en un medio libre de lipoproteínas antes de cambiarlas a un medio rico en lipoproteínas de baja densidad durante 24h. Posteriormente se fijan y tiñen con el colorante Filipin III. Ampliación a ×200
Para descartar otras enfermedades lisosomales de la misma ruta metabólica se analizó en lisado leucocitario® citario, la actividad de la lipasa ácida lisosomal13 y de la esfingomielinasa14 obteniendo valores dentro del RRI.
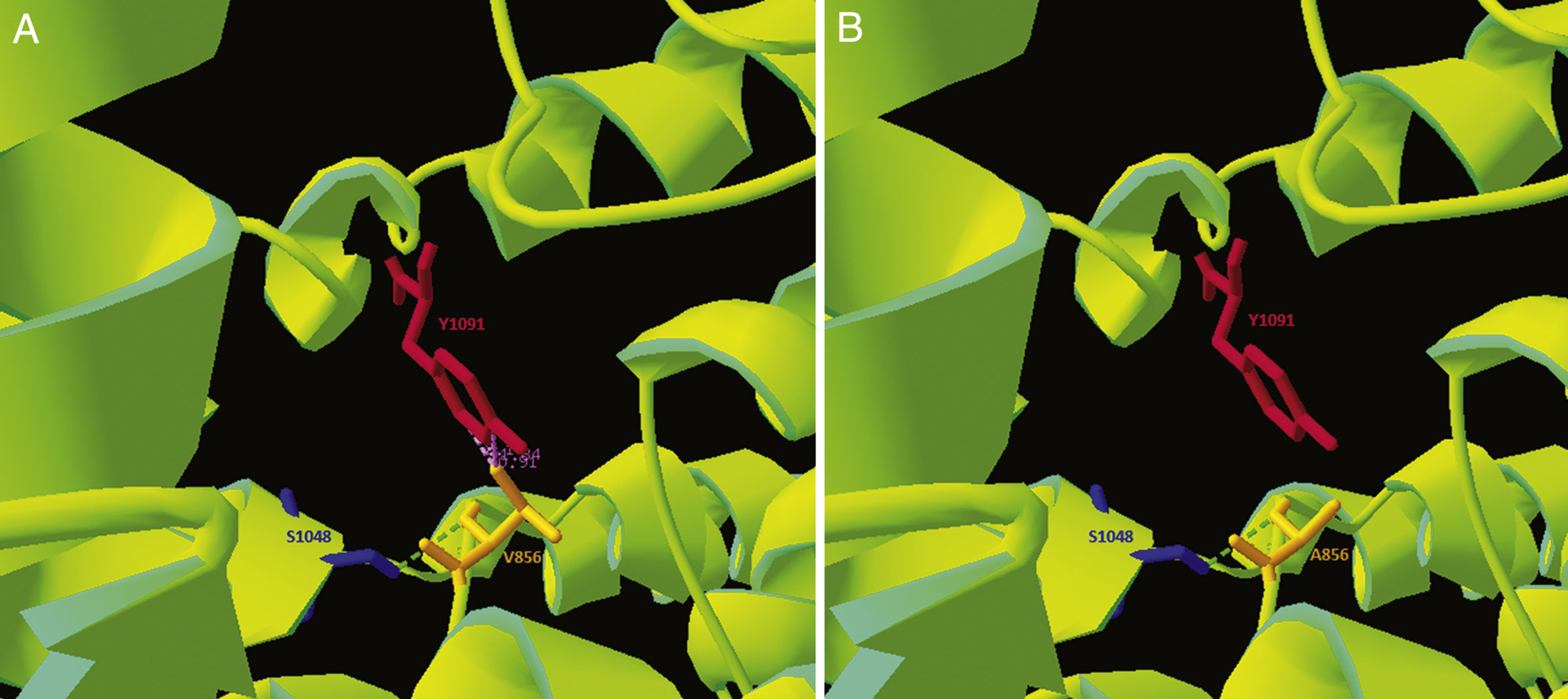
Como la variante p.Val856Ala no está descrita y se localiza en un bucle rico en cisteínas que acumula aproximadamente un tercio de las variantes patogénicas descritas1 se realizó un análisis bioinformático para la misma. Mediante Clustal Omega se realizó un análisis de conservación de la secuencia, comparándola con 30 especies, y se observó que se trataba de una región altamente conservada. Simultáneamente se utilizó la estructura de NPC1 publicada por Gong et al. en 2016 en el Protein Data Bank (Cryo-EM structure of the full-lengh human NPC1 at 4.4 angstrom; número de acceso PDB: 3JD8) y se modeló la variante patogénica con el programario libre Swiss-PdbViewer, observándose una disminución de las interacciones electroestáticas en la forma variante (fig. 2). Mediante el predictor de estabilidad DUET se observó una disminución en la estabilidad de la proteína que contenía la variante p.Val856Ala (ΔΔG: -1.457 vs. ΔΔG:0kcal/mol). Según las guías del Colegio Americano de Genética Medica15, está variante pasaría a catalogarse como altamente probable de ser patogénica.
 o Ala856 (B). Figura realizada a partir de la secuencia 3JD8 (PDB) mediante el programa Swiss PdbViewer.")
Con las evidencias presentadas en este trabajo, tanto a nivel clínico como bioquímico, molecular y celular, consideramos que se trata de un paciente de NPC por heterozigosis compuesta de la variante p.Ile1061Thr y una nueva variante asociada a patogenicidad, no descrita hasta ahora, p.Val856Ala.
Los autores quieren agradecer su colaboración en este trabajo a María Teresa Sagrario Fustero, Clementina del Canto Pérez y Adolfo Mínguez-Castellanos por su ayuda en el diagnóstico clínico del paciente, a Jorge J. Cebolla, Pilar Irún y Carmen Domínguez por su inestimable colaboración en las pruebas de laboratorio.

