



La paraplejía espástica hereditaria autosómica dominante (PEH-AD) es un grupo de enfermedades neurodegenerativas de origen genético que provocan espasticidad progresiva y debilidad en extremidades inferiores debido a la degeneración del tracto corticoespinal1. Actualmente se han descrito unos 79 loci cromosómicos y más de 70 variantes patogénicas causantes de PEH2. Un 40% de todas las familias con PEH-AD tienen variantes patogénicas del gen SPAST, en el loci 2p22.3. Dicho gen, también conocido como SPG4, está formado por 17 exones y codifica para la proteína espastina. Esta proteína es una ATPasa asociada a diversas actividades celulares como la proteólisis, el ciclo celular, el transporte vesicular, la biogénesis de peroxisomas y las funciones mitocondriales3.
Se han descrito más de 250 variantes patogénicas en el gen SPAST de diferente tipo (deleciones, inserciones y de sustitución de bases), y en la mayor parte de las familias descritas se encuentran mutaciones específicamente únicas. Algunas de estas variantes parecen condicionar determinadas particularidades como mayor precocidad y gravedad en varones, comienzo más precoz, afectación cognitiva y vesical o comienzo tardío.
El fenotipo clínico es indistinguible, entre los diferentes mecanismos mutacionales (sin sentido, deleción, reordenamiento, entre otros), siendo la haploinsuficiencia la base molecular de esta variabilidad4.
Cuando los síntomas se presentan en la infancia tienen que considerarse otros diagnósticos como lesiones estructurales, infecciones y enfermedades metabólicas. Las variantes patogénicas que producen PEH de inicio pediátrico son en el gen ATL1 (SPG3A) seguido del gen SPAST5.
Presentamos una nueva variante patogénica en el gen SPAST, en 3 miembros de una familia española afecta de PEH-AD (fig. 1).
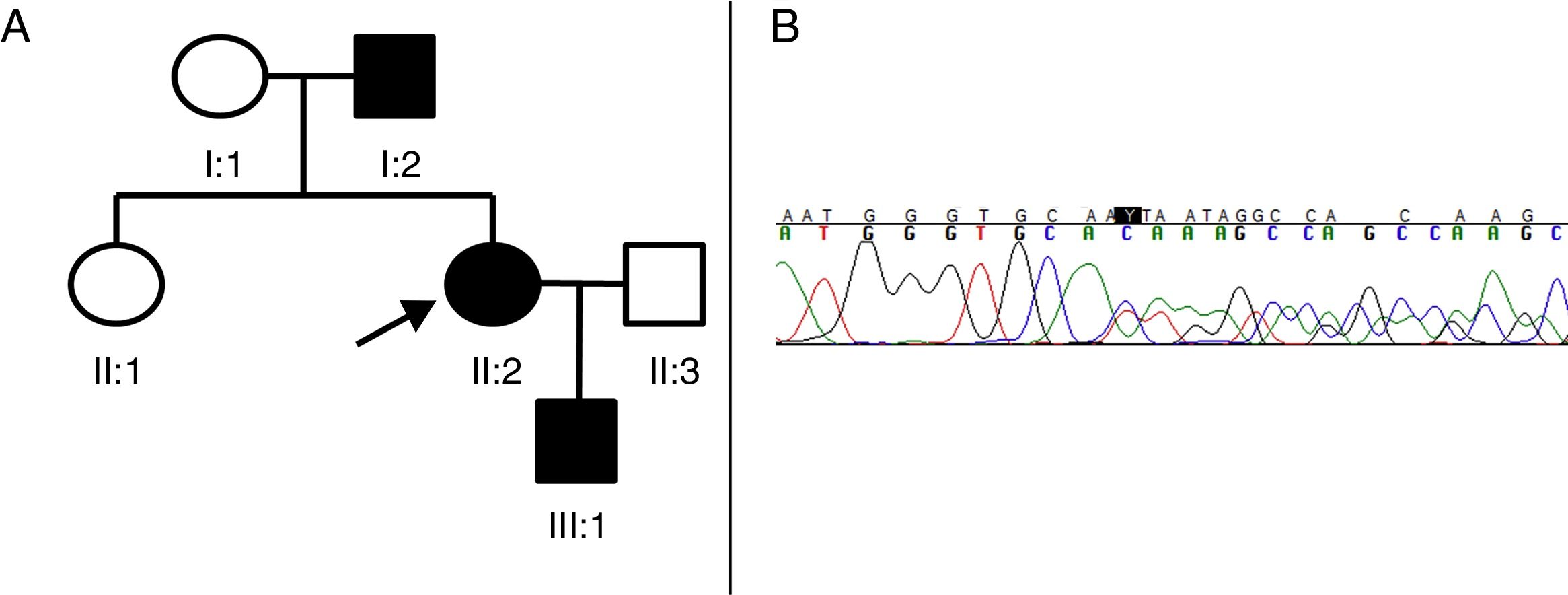
 Pedigrí de la familia afecta de PEH-AD. El caso índice (probandus) está indicado con una flecha. Los cuadrados indican los varones, los círculos indican las mujeres, los símbolos sombreados indican los pacientes afectos de PEH-AD. B) Segmento de la secuenciación del paciente II:2. Una deleción en heterocigosis de 4 nucleótidos (TGTC) se detecta en la posición c.1457 del exón 12 del gen SPAST (c.1457_1460del) que provoca un codón de parada prematuro (Thr486Ilefs*43).")
A) Pedigrí de la familia afecta de PEH-AD. El caso índice (probandus) está indicado con una flecha. Los cuadrados indican los varones, los círculos indican las mujeres, los símbolos sombreados indican los pacientes afectos de PEH-AD. B) Segmento de la secuenciación del paciente II:2. Una deleción en heterocigosis de 4 nucleótidos (TGTC) se detecta en la posición c.1457 del exón 12 del gen SPAST (c.1457_1460del) que provoca un codón de parada prematuro (Thr486Ilefs*43).
El paciente II:2 (probandus) es una mujer de 41 años, que tan solo presenta hiperreflexia y espasticidad leve en extremidades inferiores. El paciente I:2 es un varón, padre del caso índice, que empezó a desarrollar los síntomas a la edad de 73 años en forma de debilidad progresiva junto con espasticidad en extremidades inferiores, y que le provocó una incapacidad para ambular de manera autónoma en un periodo de 3 años. La neuroimagen cerebral y espinal, el estudio de líquido cefalorraquídeo (LCR) y los estudios microbiológicos (VIH, sífilis, HTLV1) no presentaron alteraciones. Finalmente, el paciente III:1 es un niño de 4 años, hijo del caso índice, que presentaba una hiperreflexia rotuliana y marcha en punta de dedos (sin signo de Babinski ni clonus) que mejoró con fisioterapia.
Considerando la posibilidad de que se tratase de una PEH de herencia dominante se decidió secuenciar el gen SPAST. Las muestras de sangre de los 3 pacientes, para el estudio del gen se obtuvo previo consentimiento informado. Los 17 exones y la región adyacente a dichos exones, del gen SPAST, se amplificaron por reacción en cadena de la polimerasa (PCR) y se analizaron mediante el análisis de polimorfismo de conformación de cadena simple (SSCP) utilizando la electroforesis capilar. Mediante la secuenciación se identificó una deleción de 4 nucleótidos (c.1457_1460del:TGTC) en heterocigosis que provocan el cambio de una treonina en isoleucina y la aparición de un codón de parada prematuro, 43 aminoácidos más tarde (Thr486Ilefs*43) (fig. 1B). Se produce una variante, clasificada como patogénica utilizando las guías de interpretación de la American College of Medical Genetics and Genomics6, por cambio de marco de lectura y que codifica una espastina truncada y disfuncional. La variante patogénica no ha sido descrita en la literatura ni en las bases de datos revisadas (Exome Aggregation Consortium, Single Nucleotide Polymorphism y NHLBI Exome Sequencing Project).
En resumen, se ha identificado una nueva variante patogénica del gen SPAST c.1457_1460del (Thr486Ilefs*43), asociada a una paraplejía espástica hereditaria con un patrón de herencia autosómica dominante. Es importante destacar, en primer lugar, que a pesar de presentar la misma variante patogénica, el fenotipo puede ser heterogéneo y la ausencia de síntomas no excluye la enfermedad, motivo por el cual se debe tener en cuenta de cara al consejo genético. En segundo lugar, recalcar el fenómeno de anticipación que resulta manifiesto entre el paciente I:2 y el paciente III:1.

