




El síndrome de Jansen-de Vries (JDVS: MIM#617450), también conocido como «trastorno del desarrollo intelectual con dificultades gastrointestinales y umbral de dolor alto (IDDGIP)», es una enfermedad autosómica dominante1,2 descrita en menos de 20 pacientes1, siendo este caso, hasta donde sabemos, el primero diagnosticado en España. Además de por su extrema rareza, su interés radica en un patrón atípico de activación sexual obsesiva, y en la ausencia de discapacidad intelectual.
El gen PPM1D es un miembro de la familia PP2C de proteínas fosfatasas serina/treonina, que codifican la proteína fosfatasa Mg2+/Mn2+ dependiente de 1D. Este gen participa en la regulación negativa del estrés celular dependiente de p533. Jansen et al. fueron los primeros en identificar mutaciones de novo de tipo truncante o frameshift en los exones 5 y 6 del gen PPM1D2, como responsables de un síndrome que se caracteriza por un fenotipo peculiar, discapacidad intelectual, afectación del lenguaje y de la conducta, dificultades gastrointestinales con períodos de vómitos2, elevada sensibilidad para los sonidos y baja para el dolor2,4.
Se trata de un varón de 8 años evaluado previamente por retraso psicomotor leve (marcha liberada con 2 años), y por un intenso retraso del lenguaje y la comunicación (primeras palabras con 4 años).
El embarazo cursó con crecimiento intrauterino retardado armónico. El parto fue eutócico a las 39 semanas. Apgar 9/10, peso: 2.175kg (percentil 1), longitud 47cm (percentil 3) y perímetro cefálico 33cm (percentil 5). Destacó inicialmente la presencia de hipotonía, dificultades para la alimentación, reflujo gastroesofágico y estreñimiento, que a lo largo de los años alternó con diarreas intermitentes.
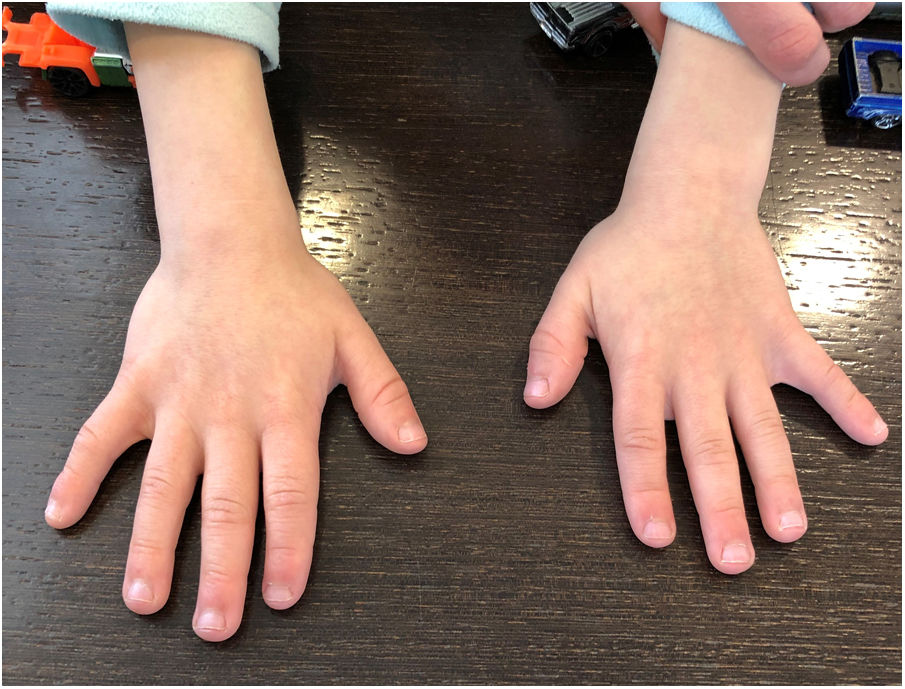
La exploración física con 7 años mostraba un peso de 20,4kg (percentil 13), talla de 112cm (percentil 2) y perímetro craneal de 48,5cm (percentil 3). Se observaban discretas dismorfias descritas en la figura 1. Además, mostraba hiperlordosis lumbar, manos pequeñas con braquidactilia (sobre todo de falanges distales) y clinodactilia (fig. 2) y pies pequeños, con displasia ungueal bilateral. Durante la exploración neurológica no se apreciaron signos de focalidad.
, leve hipotelorismo, epicanto bilateral de predominio izquierdo, nariz bulbosa con narinas pequeñas, filtro liso, labio superior fino e inferior grueso y boca grande.")
Fenotipo del paciente a la edad de 7 años. Frente amplia, pabellones auriculares de implantación baja, amplios y rotados posteriormente (se objetiva el derecho en la foto), leve hipotelorismo, epicanto bilateral de predominio izquierdo, nariz bulbosa con narinas pequeñas, filtro liso, labio superior fino e inferior grueso y boca grande.
 y clinodactilia.")
Había una preocupación ambiental por una conducta sexual desinhibida, caracterizada por un intenso deseo de relaciones sociales (a pesar de ciertas dificultades para adaptarlas a sus compañeros) y obsesiones de contenido erótico, asociado a un onanismo compulsivo. Existía un elevado umbral para el dolor, hipersensibilidad a los sonidos y una estereotipia de «descarga» con ambas manos a la altura de la cara. Los comportamientos repetitivos no eran invalidantes y su contacto ocular y empatía eran buenos. En la evaluación neuropsicológica, el WISC-V destacó por un cociente intelectual total de 84, con una memoria de trabajo de 74 como ítem más afectado. Además, se comprobaron dificultades atencionales marcadas.
El estudio de edad ósea objetivaba un retraso de 2 años −3DS−. Los resultados de la exploración cardiológica, la RM cerebral y el EEG fueron normales. El estudio se amplió mediante secuenciación completa del genoma en trío (WGS), que mostró una mutación de novo, frameshift, en el exón 5, en heterozigosis, del gen PPM1D (hg19; chr 17: 58734146; NM_003620.3; c.1206_1207del, p.Asn402Lysfs*31). Esta mutación, confirmada posteriormente por Sanger, no ha sido previamente descrita en las bases de datos ni en la bibliografía médica.
Las características físicas más frecuentemente observadas en el JDVS son la hipotonía2, la edad ósea retrasada4, las dificultades de alimentación y los problemas gastrointestinales (incluidos los vómitos y el estreñimiento)1–4, y dismorfias como las anormalidades faciales leves comentadas2,3, la microcefalia, la hiperlordosis4, las manos pequeñas con braquifalangia (detectables en el 90% de los casos) y uñas algo hipoplásicas1–4. Dentro de las dificultades cognitivo-comportamentales, las más frecuentes son el retraso en el habla4, el TDAH2 y la ansiedad con componente rígido-obsesivo2,3 acompañada de hiper o hiporreactividad a los estímulos sensoriales (hipersensibilidad al sonido3, umbral alto al dolor en el 90% de los casos)2–4; muchas de ellas se encuentran en nuestro paciente. La hipersexualidad ha sido descrita previamente en pacientes con discapacidad intelectual y puntualmente en pacientes con trastornos del espectro autista5. Actualmente no se conoce si dicha hiperactivación forma parte de un fenotipo conductual específico, escasamente referido con anterioridad.
Destacamos la presencia del JDVS en un caso con dismorfias características, pero sin discapacidad intelectual y con una curiosa y llamativa conducta hipersexual, secundario a una mutación no descrita previamente. Si bien la discapacidad en grado variable ha sido la norma en este síndrome1, Jansen et al., en su serie, describió otro caso con JDVS sin discapacidad intelectual2, mostrando la necesidad de estudios genéticos ampliados ante la presencia de trastornos del neurodesarrollo complejos de causa desconocida.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.

