



Diferentes estudios han asociado la deficiencia en VD a la esclerosis múltiple, lo que ha llevado a plantear su potencial papel en la respuesta inmunitaria. Existe menos información sobre su papel en la remielinización.
DesarrolloEn las células del SNC existe el receptor VD, así como las enzimas que transforman los metabolitos de la VD para poder activar este receptor, lo que plantea un potencial efecto de la VD. Tanto estudios in vitro como modelos animales han mostrado que la VD puede tener un papel sobre la mielinización actuando en factores que influyen en el microambiente que favorece la mielinización como en la proliferación y la diferenciación tanto de las células madre neuronales en células precursoras de oligodendrocitos como en estas en oligodendrocitos. No se conoce si los mecanismos de internalización de la VD en el SNC son sinérgicos o antagónicos a los que permiten la entrada de los metabolitos de la VD en las células inmunitarias.
ConclusionesLa VD debe tener un papel en el SNC y se puede hipotetizar si actúa en la remielinización. El conocimiento de los mecanismos básicos de los efectos de la VD en la mielinización parece necesario para poder aconsejar a los pacientes con esclerosis múltiple ante deficiencias de VD en la clínica.
Several studies have found an association between multiple sclerosis and vitamin D (VD) deficiency, which suggests that VD may play a role in the immune response. However, few studies have addressed its role in remyelination.
DevelopmentThe VD receptor and the enzymes transforming VD into metabolites which activate the VD receptor are expressed in central nervous system (CNS) cells, which suggests a potential effect of VD on the CNS. Both in vitro and animal model studies have shown that VD may play a role in myelination by acting on factors that influence the microenvironment which promotes both proliferation and differentiation of neural stem cells into oligodendrocyte progenitor cells and oligodendrocytes. It remains unknown whether the mechanisms of internalisation of VD in the CNS are synergistic with or antagonistic to the mechanisms that facilitate the entry of VD metabolites into immune cells.
ConclusionsVD seems to play a role in the CNS and our hypothesis is that VD is involved in remyelination. Understanding the basic mechanisms of VD in myelination is necessary to manage multiple sclerosis patients with VD deficiency.
La vitamina D (VD) es un grupo de hormonas, que incluyen la VD 2 o ergocalciferol y la VD 3 o colecalciferol. La VD se adquiere principalmente por la exposición de la piel a la luz solar y por la ingesta alimentaria. La existencia de diferentes estudios de epidemiología analítica ha permitido establecer la posibilidad de una relación entre la esclerosis múltiple (EM) y la deficiencia de VD, que ha sido motivo de reflexión entre los investigadores1-6. Esta asociación podría ser debida a mecanismos causales, a interacción genético-ambiental o simplemente a la conjunción con los muchos factores ambientales que se observan en la EM, pero no se comprende cuál es la base biológica de esta relación. Para tratar de conocerlos muchos investigadores han centrado su atención en el papel de la VD y sus metabolitos en estudios in vitro y en la encefalomielitis alérgica experimental (EAE)7,8. Aunque la mayoría de estas investigaciones han analizado la relación entre la VD y el riesgo de inflamación, algunos autores han evaluado el potencial papel de la VD en la mielinización y la remielinización9, motivo de esta revisión.
La definición de deficiencia de vitamina DLa determinación plasmática de 25(OH)D es el marcador que mide habitualmente la suficiencia de VD, debido a que este metabolito tiene una vida media plasmática larga (15-35 días). Sin embargo, en los últimos años se ha cuestionado que la determinación total de 25(OH)D suponga una demostración adecuada del nivel de suficiencia de VD. Esta discusión se basa en la determinación de formas activas o de las fracciones libres de las hormonas e indicaría que, de forma similar a otras hormonas, aquellos metabolitos que están unidos a proteínas podrían ser inactivos y no representarían un marcador adecuado10. La VD circula asociada a una proteína específica, a albúmina y de forma libre. La proteína específica es la proteína de unión de la VD (DBP). La fracción libre de la VD representa una parte muy pequeña de los metabolitos circulantes, algo menos del 1%, y puede tener una función biológica diferente de la fracción ligada a proteínas11,12. Bajo estos parámetros, los conceptos de suficiencia o deficiencia de VD no son fáciles de definir porque, además, la concentración plasmática de 25(OH)D circulante varía dependiendo de distintos estados fisiológicos o patológicos y bajo la influencia de factores genéticos13. Para añadir más complicación a la definición del estado del nivel circulante de VD en un sujeto determinado, el cálculo de la fracción libre se realiza habitualmente a través de estimaciones matemáticas y no por medición directa14 y esta forma de calcular la fracción libre está bajo discusión, ya que cuando se ha comparado con la determinación directa se ha observado que existen diferencias destacables. Existe una cuestión terminológica en la literatura que hay que tener en cuenta al analizarla para evitar confusiones. Así, se ha denominado como 25(OH)D «biodisponible» a todo el 25(OH)D circulante que no está unido a DBP, es decir, la fracción libre y la fracción unida a albúmina, y representa aproximadamente el 10% de todo el 25(OH)D circulante, que no debe confundirse con la 25(OH)D libre. Estos aspectos metodológicos hacen que no sea fácil establecer el papel de la deficiencia de VB como riesgo y todavía más difícil personalizarla en individuos.
Transporte plasmático de la vitamina D y transformación a metabolitos activosComo se ha señalado, la VD utiliza proteínas transportadoras, DBP o albúmina, que permiten que acceda a los tejidos y células que la necesitan. La DBP se produce en el hígado y además del transporte participa en la liberación del metabolito con el objetivo de facilitar la conversión de la prohormona 25-hidroxivitamina D (25[OH]D), que es circulante pero no es activa, al metabolito 1,25-dihidroxivitamina D (1,25[OH]2D), que es el metabolito activo. Para ello, es necesaria la actuación de la 25-hidroxi-vitamina D-1α-hidroxilasa, que también es conocida como la citocromo p450 27B1 (CYP27B1) o como 1α-hidroxilasa y es codificada por el gen CYP27B1. Esta enzima está localizada en el túbulo renal y en otros tipos celulares, como células inmunitarias y del sistema nervioso central (SNC), y cataliza la hidroxilación de 25(OH)D a 1,25(OH)2D. Este metabolito actuará sobre el receptor nuclear de 1,25(OH)2D, el receptor de VD (VDR). Como ya se ha señalado previamente, el 99% de 25(OH)D circula unida a DBP o a la albúmina15, mientras que solo una existe una pequeña parte que circula de forma libre16. Debido a que 25(OH)D es lipófila, tiene la capacidad de difundirse de forma pasiva a través de las membranas celulares. La idea de que la fracción libre, como ocurre con otras hormonas, es la forma activa está muy cuestionada. Primero, porque las concentraciones de la fracción libre de 1,25(OH)2D en el suero son aproximadamente 10–13M17 más bajas (casi 1.000 veces) que las necesarias para la unión con VDR. Segundo, por el importante papel que se ha demostrado de la megalina, también denominada low-density lipoprotein-related protein 2 (LRP2). Esta proteína transmembrana actúa como un receptor multiligando en varios tejidos y se une a la BDP generando un compuesto que será internalizado a través de endocitosis en las células del túbulo renal proximal, donde se halla la 1α-hidroxilasa. La megalina actúa como un receptor endocítico que se une a ligandos extracelulares para poder internalizarlos mediante endocitosis, tras lo cual se activan determinadas vías de señalización que afecta a lipoproteínas, hormonas, fármacos y a la propia DBP, y que dependen de la unión con unas proteínas adaptadoras que reconocen determinadas zonas citoplasmáticos de megalina. Las funciones donde estas vías actúan influyen en el tráfico de proteías18, la interacción con proteínas del citoesqueleto o citoplasmática o su participación en la señal SHH19. La megalina puede ser fosforilada por GSK3, PKC, CK- II o PKA para su reciclaje desde los endosomas20. Los ratones deficientes para megalina desarrollan un fenotipo que se asemeja en el hueso al raquitismo que se observa en los ratones deficientes en VD21. Junto a la megalina, hay 2 proteínas más que actúan conjuntamente en la unión a DBP, que son la cubulina22 y la proteína adaptadora Dab223. Aparte del riñón, la megalina se expresa en otros tejidos, como placenta, glándula mamaria y las glándulas paratiroides, por lo que se ha indicado que este mecanismo sea el que utiliza VD en estos tejidos y también está presente en el sistema nervioso.
En el cerebro sano, se ha descrito que la megalina se ubica preferentemente en las células ependimarias que recubren la pared ventricular, los capilares y el plexo coroideo24, pero también se ha hallado en el SNC y en el SNP. La deficiencia en el gen que codifica la megalina produce una alteración en el desarrollo del cerebro anterior, con la ausencia de aparato olfativo y malformaciones cráneo-faciales25 y una mutación en este gen produce anormalidades en el diencéfalo dorsal con más hipertrofia del plexo coroideo en el tercer ventrículo26. Dentro del SNC, lamegalina se expresa en neuronas y astrocitos27. En astrocitos, la megalina es necesaria para poder internalizar albúmina que es requerida para las señales que conducen a la síntesis de factores neurotróficos por neuronas vecinas28. También se ha identificado en las células ganglionares de la retina. En estas células, interactúa con la metalotioneína-IIA permitiendo la activación de diferentes vías de señalización intracelular implicadas en la neuroprotección29. En muestras de cerebro de seres humanos sanos, monos, cerdos, ratas y ratones se demostró la expresión de megalina en diferentes poblaciones neuronales de la corteza cerebral, el hipocampo, el cuerpo estriado, el tálamo, el bulbo olfativo y el cerebelo. Se ha observado que la megalina está sobrerregulada en neuronas afectadas en la AD30. En el cerebro, la megalina participa en la endocitosis e internalización de apolipoproteína E y la proteína precursora de amiloide (APP).
Internalización de los metabolitos de la vitamina DEl mecanismo de unión DBP-megalina es la forma de internalizar los metabolitos de la VD en muchos tejidos y, si la fijación a la DBP es muy alta, habrá una mayor cantidad de metabolitos de la VD disponibles para ello y una menor fracción libre que pueda internalizarse a través de una difusión pasiva. De manera que el nivel de DBP influye en la disponibilidad de fracción libre de VD y, en consecuencia, un aumento de niveles circulantes de VD puede suponer una menor fracción libre disponible. Por el contrario, una menor concentración de DBP podría aumentar las concentraciones de metabolitos «libres» de VD en la membrana celular, lo que facilitaría la difusión pasiva de estas moléculas. Este concepto no es tan estricto porque podría ser posible que para algunas células hubiera mecanismos de internalización de DBP con la unión a otras proteínas que no sean la megalina y que internalizaran los metabolitos de VD fijados y no libres. En este sentido, la absorción de 25(OH)D en los linfocitos B se puede realizar desde la molécula fijada a DBP al margen de la megalina y también a través de la unión de DBP a proteínas receptores Fc gamma, que también se asocian a inmunoglobulinas. A pesar de ello, hay tejidos que no expresan megalina, lo que indica que en estos tejidos se internaliza la 25(OH)D libre. Esta posible actuación de la 25(OH)D libre se plantea especialmente en las células inmunitarias, como monocitos, macrófagos y células dendríticas, entre otros. Estudios in vitro han demostrado que los monocitos expuestos a dosis crecientes de 25(OH)D en presencia de DBP muestran inducción dependiente de la dosis de las proteínas antibacterianas, pero, en ausencia de DBP, esta respuesta es mucho más potente y, en consecuencia, la capacidad de 25(OH)D para promover la actividad antibacteriana de monocitos es dependiente tanto de la concentración de suero y el genotipo de DBP31. Observaciones similares también se han observado con células T reguladoras. Sin embargo, esta relación inversa está influida por otras variables. Así, los metabolitos de la VD pueden unirse también a la albúmina de suero, aunque la afinidad de 25(OH)D y 1,25(OH)2D para la albúmina es sustancialmente menor; además, debido a la abundancia relativa de la albúmina en el suero (650mM) en comparación con DBP (5mM), debe tenerse en consideración la posibilidad que en determinadas situaciones los metabolitos de VD puedan ser transportados por la albúmina y es posible que parte de la DBP en el suero esté vacía, de manera que la fijación de 25(OH)D y 1,25(OH)2D a la albúmina también influiría en la fracción libre. En el caso de 25(OH)D se estima que menos del 0,1% de los niveles circulantes totales de este metabolito son la fracción libre. Un segundo aspecto es que modificaciones dietéticas pueden influir en la afinidad de la DBP por los metabolitos de VD, como, por ejemplo, los ácidos grasos poliinsaturados que disminuyen la afinidad de unión y, por lo tanto, modificar su biodisponibilidad. En este sentido, no se conoce el comportamiento de la 25(OH)D libre en pacientes que reciben suplementos de VD tanto por vía oral como parenteral y es importante puesto que, teniendo en cuenta que las concentraciones séricas de 25(OH)D correlacionan con la concentración de DBP, puede que estos suplementos no modifiquen la concentración de 25(OH)D ni total ni libre. Así, se ha descrito que las concentraciones séricas totales de 25(OH)D, 1,25(OH)2D y DBP tras suplementos con VD3 o VD2 no modifican al inicio del tratamiento, aunque al final del mismo los niveles séricos de 25(OH)D3 aumentan significativamente más que los de 25(OH)D2, modificándose también la concentración de DBP en el suero, pero en cambio los niveles de 25(OH)D libre o 25(OH)D biodisponible fueron similares tanto con suplementos de VD 2 y/o VD 3. La menor eficiencia de la VD 2 para elevar los niveles séricos totales de 25(OH)D frente a la VD 3 se ha atribuido a que 25(OH)D2 tiene una menor afinidad para unirse a DBP en relación con 25(OH)D3.
Un tercer factor que influye en la relación DBP con la fracción libre es que la DBP presenta variaciones fenotípicas que influyen en su afinidad con los metabolitos de VD. Estas variaciones representan que la DBP está constituida por proteínas que tienen algunas diferencias y que están relacionadas con el componente del grupo-específico (Gc) 1f, Gc1s y Gc2, y que tienen que ver con polimorfismos en el gen de la DBP o gen Gc. Estas variaciones se producen por secuencia de aminoácidos distintos de la DBP (Gc1s, Gc1f, y Gc2) que alteran la afinidad de unión a los metabolitos de VD para la DBP, de forma que aquellos que muestran Gc1f tienen la más alta avidez y los de Gc2 la más baja32,33, y ello se compensa con variaciones en los niveles séricos de DBP siendo el Gc2 el menos alto y el Gc1f el más alto34, es decir, a menos afinidad, mayor concentración de DBP y viceversa. Esta variabilidad en los alelos Gc se asocia a diferencias raciales, de forma que las poblaciones de razas negras y asiáticas muestran una mayor presencia de la forma Gc1f de DBP, mientras que la raza blanca muestra con mayor frecuencia la forma Gc1s. La forma Gc2, que tiene menor afinidad por la DBP, se encuentra más en la raza blanca y rara vez se encuentra en la raza negra, de manera que se ha indicado que estas diferencias de afinidad reflejan la mayor pigmentación de la piel. Dentro de las variaciones fenotípicas de la DBP, pueden a su vez producirse cambios derivados de polimorfismos (SNP), como rs7041 y rs4588, entre otros. Los polimorfismos rs4588 y rs7041 están asociados con los niveles de 25(OH)D más bajos y se observan en mayor frecuencia en hispánicos y afroamericanos35. Las combinaciones de estos alelos pueden alterar la concentración de DBP y la afinidad para 25(OH)D y 25(OH)36,37. Así, Santos et al. han encontrado que los polimorfismos rs4588 y rs7044 están asociados con concentraciones de 25(OH)D más bajas en mujeres jóvenes y sanas38. Cheung et al. encuentran 4 polimorfismos (rs2282679, rs10741657, rs12785878 y rs6013897) y describieron que mientras rs2282679 está asociado con los niveles séricos de 25(OH)D bajos, rs12785878 está asociado solo con deficiencia de VD39.
El concepto de que la internalización en la mayoría de las células inmunitarias es independiente de la megalina es importante, porque implica que el acceso de los metabolitos de la VD a estas células podría depender o de la unión a receptores Fc gamma o de la fracción libre de la VD, que tiene una relación inversa con las concentraciones plasmáticas de DBP y, en consecuencia, a las concentraciones plasmáticas circulantes de VD, y es la situación opuesta a las células del SNC que sí utilizan la internalización por megalina. De esta forma, podría hipotetizarse que un nivel bajo de DBP podría suponer una internalización alta a las células inmunitarias y opuesta a las células del SNC.
Las enzimas que participan en el metabolismo de la vitamina DVarias enzimas participan específicamente en el metabolismo de la VD40. La primera es CYP2R1, que convierte la VD en 25(OH)D41. Una de ellas, 1α-hidroxilasa (CYP27B1), que es codificada por el gen CYP27B1, que ha sido citada previamente, cataliza la hidroxilación de 25(OH)D a 1,25(OH)2D, que se unirá a el VDR. La segunda es la 1,25-dihidroxivitamina D 24-hidroxilasa (24-hidroxilasa, CYP24A1), que es una enzima que es codificada por el gen CYP24A1 y actúa en la degradación a través de la hidroxilación de 1,25(OH)2D y, en consecuencia, actúa en el camino opuesto. Las 2 corresponden a enzimas de la superfamilia de citocromo p450. No se hallado relación de variantes en el gen que codifica la DBP con la EM42.
El receptor de vitamina DEl 1,25(OH)2D es el metabolito de la VD que se une al VDR, que es un receptor nuclear. En situaciones patológicas, el VDR puede distribuirse principalmente en el citoplasma43. Cuando se produce la interacción del VDR con su ligando 1,25(OH)2D se produce la formación de 2 superficies de interacción donde participan proteínas independientes e interactúa con el receptor retinoide X (RXR) necesario para la unión con el ADN, que se requiere para el reclutamiento de correguladores necesarios para la modulación genética44. La unión 1,25(OH)2D-VDR se dimeriza con RXR y se transloca al núcleo donde se une a elementos de respuesta a la VD (VDRE) que actúan con los genes de respuesta a la VD. Los genes diana pueden ser coactivadores o correpresores y a través del complejo VDR/RXR se produce una inducción o represión de la transcripción en el que participan ATPasas y proteínas involucrados en el reclutamiento de la ARN polimerasa ii45. Aparte de este mecanismo relacionado con VDRE, VDR puede inhibir genes antagonizando ciertos factores de transcripción46,47. Existen variantes alélicas del gen VDR. Se ha descrito una mayor susceptibilidad de determinadas variantes a las infecciones y una mayor incidencia de enfermedades autoinmunes, como la EM y el cáncer, y ello podría ser debido a una reducción de la estabilidad VDR-mRNA y, por lo tanto, a la reducción de expresión de VDR. Existen algunas variaciones del gen VDR que conduce a un receptor no funcional48. Se han descrito polimorfismos en el gen que codifica VDR (ApaI, TaqI, FokI and BsmI) y se ha encontrado estudios con asociaciones significativas a la EM49-51 y otros no52-57.
Metabolismo de la vitamina en el sistema nervioso centralAunque tradicionalmente se ha considerado que la VD participa en el metabolismo óseo, su presencia en el SNC indica que actúa en algunas de sus funciones, incluso es considerada como un neuroesteroide58. La 25(OH)D se encuentra en el LCR de pacientes con EM y en los sujetos de control y se correlaciona positivamente con 25(OH)D sérico59, y los niveles de DBP también están presentes en el LCR de controles y son más prevalentes en los pacientes con EM60. Esto confirma que la VD y su principal proteína transportadora acceden al SNC. En la EAE, se ha demostrado un aumento en la expresión de mRNA-VDR y de mRNA-CYP27B1 en el SNC frente a controles61. Teniendo en cuenta que la expresión de VDR y CYP27B1 está presente en neuronas y astrocitos de donantes sanos, se podría formular la hipótesis que la VD puede tener acciones específicas sobre el SNC62. En este sentido, ha sido descrito que en cultivos in vitro de células gliales suplementados con 1,25(OH)2D3 la glía muestra efectos inflamatorios63, lo que apoyaría un potencial terapéutico en la EM. Considerando que 1,25(OH)2D puede atravesar la barrera hematoencefálica y que la mayoría de las células del SNC, incluyendo la microglía64, expresan VDR, la idea de una acción directa sobre el SNC de la VD es una posibilidad y recientemente se ha señalado la acción de la VD sobre las histonas generando una alteración epigenética65, que ha sido relacionada con la EM66,67. Como se ha descrito, VDR se hallan presentes en neuronas y glía68,69. VDR y CYP27B1 están expresados en neuronas y astrocitos de personas sanas62. En ratas hembras con EAE, se observó que 1,25(OH)2D3 incrementa la expresión de nRNA-VDR y mRNA-CYP27B1 en comparación con los controles61. Se produce un aumento de la expresión de mRNA-CYP24A1 por la exposición a la 1,25(OH)2D en el cultivo de la línea celular astrocitoma C6 y en el cultivo de astrocitos procedentes de rata70. La expresión de CYP24A1 en macrófagos es modificada por IFN-γ71. Se ha observado asimismo una reducción de la producción de TNF y de interleucina-1β en una línea celular de microglía expuesta a 1,25(OH)2D72 y una reducción de nRNA-TNF y del mRNA del factor estimulante de colonias de macrófagos en cultivo células primario de astrocitos de rata y en una línea celular de astroglioma73. Asimismo la inmunorreactividad de MHC clase disminuye en la EAE después del tratamiento con 1,25(OH)2D74.
Es especialmente interesante el artículo de Smolders et al.75 porque permite extrapolar la presencia del metabolismo de la VD en el SNC sano y en la EM. Estos autores confirman la expresión nuclear de VDR en todas las neuronas, incluso en hipotálamo. La CYP24A1 está expresada en el citoplasma de prácticamente todas las neuronas, incluido el hipotálamo, pero sobre todo en los núcleos supraóptico y periventricular, y en el hipotálamo colocaliza con cortisol-releasing hormone (CRH), vasopresina y oxitocina. En la sustancia blanca normal, en pacientes y en controles se halla la actividad nuclear de VDR en oligodendrocitos (OL). También hay expresión de VDR en la microglía, pero no de CYP24A. En astrocitos se observa tanto la expresión de VDR nuclear como CYP24A citoplasmática. Sin embargo, en algunos pacientes se halla la expresión de VDR en citoplasma, especialmente en aquellas células gliales que tienen mayor expresión de GFAP. En la sustancia blanca de apariencia normal, la expresión glial de mRNA-VDR y de mRNA-CYP27B1 no varía frente a lo que se halla en células mononucleares periféricas, mientras que la expresión de mRNA-CYP24A1 y mRNA-LRP2 están aumentadas en los controles. En la EM, la expresión de mRNA-VDR está aumentada significativamente frente a controles, mientras que las expresiones de mRNA-CYP24A1, mRNA-CYP27B1 y mRNA-megalina no varían. Asimismo estos autores hallan expresión de VDR citoplasmático en la glía en el centro de las lesiones de EM crónicas no activas y en algunas células hay un anillo perilesional con expresión de VDR. La expresión de mRNA-VDR y mRNA-CYP27B1 es mayor en las lesiones crónicas que muestran actividad frente a la sustancia blanca a normal. No hay diferencias, sin embargo, entre las lesiones crónicas no activas con la sustancia blanca aparentemente normal. La expresión de mRNA-megalina está disminuida en las lesiones crónicas activas o no frente a la sustancia blanca aparentemente normal.
Smolders et al.75 incluyen en su artículo una serie de experimentos in vitro que muestran el efecto de la adición de VD a determinadas líneas celulares relacionadas con el SNC. Así, la adición in vitro de 1,25(OH)2D3 en un cultivo con la línea celular SH-SY5Y de neuroblastoma, que incluye básicamente neuronas, induce la expresión de mRNA-CYP24A. La adición de 1,25(OH)2D3 a cultivos in vitro a una línea celular de astrocitos humanos primarios y a líneas celulares de astroglioma (U343 y U373) mostraron la sobrerregulación dependiente de la dosis de mRNA-VDR y de mRNA-CYP24A1 pero no se produjo un efecto sobre la expresión de mRNA-CYP27B1. En un cultivo primario de astrocitos y en un cultivo de microglía se añadieron IFN-γ, TNF y 1,25(OH)2D3. El IFN-γ y el TNF sobrerregularon mRNA-CYP27B1 en la microglía y los astrocitos. Tanto en las células tratadas con IFN-γ y TNF, la adición de 1,25(OH)2D3 supuso que el incremento en la expresión de mRNA-CYP27B1 fue menor que sin añadir 1,25(OH)2D3. La sobrerregulación de CYP27B1 y CYP24A1 producida por IFN-γ/TNF y 1,25(OH)2D3 fue más pronunciada en astrocitos que en microglía. A través de estudios inmunohistoquímicos han localizado megalina en el endotelio de la microvasculatura cerebral y el plexo coroideo76 y, por tanto, debe plantearse que la DBP y la VD acceden al SNC a través de la megalina y se ha hallado que la expresión de mRNA-megalina está disminuida en las lesiones de MS, tanto activas como inactivas. El mecanismo por el cual 1,25(OH)2D favorece la diferenciación neuronal no está claro, pero probablemente sea a través de actuar sobre factores de crecimiento, habiéndose observado cómo 1,25(OH)2D sobrerregula la expresión de NT-3 y NGF77, así como GDNF78, CNTF o BDNF79.
Metabolismo de la vitamina D y remielinización en la esclerosis múltipleLa mielinización es el proceso que conduce a dotar a axones mielinizables de una envoltura de mielina. Hay evidencia de que al inicio de la EM existe una remielinización en las lesiones desmielinizadas80-82, lo que también se observa en neuroimagen83-89 pero es incompleta90 y desaparece con el tiempo91, y ello puede producirse debido a la dificultad de migración y acceso de las células precursoras de OL (OPC) a las áreas desmielinizadas92, o a que en ellas no se encuentran las condiciones adecuadas para su diferenciación93. La posibilidad que ello sea producido por un agotamiento del número de las OPC disponibles para la remielinización con el tiempo no parece que sea posible porque los estudios post mortem de pacientes con EM revelan que ello no es así94-97. La remielinización disminuye con la edad al margen de cualquier enfermedad98,99. Por ello, lo más probable es que las OPC no sean capaces de madurar en OL, porque existe un microambiente que evita la diferenciación de las OPC y la posterior remielinización de los axones. Todo ello es relevante en el tratamiento de los pacientes con EM, ya que la actual terapéutica solo es efectiva en el control de los mecanismos inmunitarios y, en consecuencia, en las fases iniciales de la enfermedad, pero no tienen acción en la remielinización.
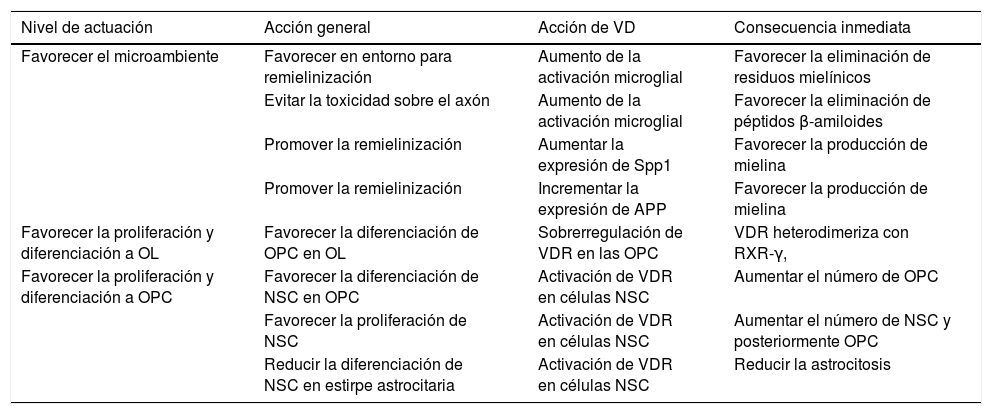
Se sabe poco sobre el papel de la VD durante la mielinización, pero varios estudios han indicado un potencial papel de la VD en la mielinización y la remielinización100–108, a diferentes niveles (tabla 1). La asociación de componentes mielínicos y VD protege con la EAE103,104 y un modelo de enfermedad de Krabbe105. Dado que la VD disminuye la expresión de la sintasa del ácido nítrico inducible en la microglía, podría influir en el equilibrio inflamatorio-antiinflamatorio que es relevante en la remielinización. La VD también aumenta la activación microglial, lo que podría facilitar la eliminación de los restos de mielina y facilitar la remielinización106. Si se añade VD a un cultivo de células OPC se induce una sobrerregulación de la transcripción de VDR, así como de NGF, pero no del mRNA de proteína básica de mielina o la proteína proteolipídica. Como se ha señalado previamente, los OL expresan VDR y la depleción de 1,25(OH)2D conduce a una disminución de la diferenciación hacia OL y desmielinizacion107, y por ello la VD y VDR son reguladores positivos de la diferenciación de las OPC. La expresión de VDR ha sido descrita en las OPC en cultivo en el tejido de la EM108,109 En las células OPC, VDR heterodimeriza junto a RXR-γ y participa en la diferenciación OPC que se expresa en las OPC durante la remielinización110. El bloqueo de VDR reduce la diferenciación OPC in vitro111 y la mielinización y la remielinización, y la activación de VDR a través de VD aumenta la diferenciación. Asimismo, las células madre de origen neural (NSC) expresan VDR y 1,25(OH)2D. 1,25(OH)2D incrementa la proliferación de las NSC y su diferenciación a neuronas y OL reduciendo la artrogliosis112. La administración de VD promueve la proliferación de células NSC113. La Spp1 es una citocina regulada por la VD y ha sido implicada en la EM114, mejora la formación de la mielina in vitro115 y se expresa en niveles altos durante la remielinización en un modelo animal de la toxina inducida por la desmielinización116.
Niveles en el proceso de remielinización donde podría actuar la VD
| Nivel de actuación | Acción general | Acción de VD | Consecuencia inmediata |
|---|---|---|---|
| Favorecer el microambiente | Favorecer en entorno para remielinización | Aumento de la activación microglial | Favorecer la eliminación de residuos mielínicos |
| Evitar la toxicidad sobre el axón | Aumento de la activación microglial | Favorecer la eliminación de péptidos β-amiloides | |
| Promover la remielinización | Aumentar la expresión de Spp1 | Favorecer la producción de mielina | |
| Promover la remielinización | Incrementar la expresión de APP | Favorecer la producción de mielina | |
| Favorecer la proliferación y diferenciación a OL | Favorecer la diferenciación de OPC en OL | Sobrerregulación de VDR en las OPC | VDR heterodimeriza con RXR-γ, |
| Favorecer la proliferación y diferenciación a OPC | Favorecer la diferenciación de NSC en OPC | Activación de VDR en células NSC | Aumentar el número de OPC |
| Favorecer la proliferación de NSC | Activación de VDR en células NSC | Aumentar el número de NSC y posteriormente OPC | |
| Reducir la diferenciación de NSC en estirpe astrocitaria | Activación de VDR en células NSC | Reducir la astrocitosis |
La activación microglial favorecida por la VD produce la fagocitosis de los péptidos β-amiloides (Aβ)117 evitando el daño sobre el axón ya que en las placas de desmielinización existe un aumento de la expresión de Aβ118-120, que inicialmente es favorecedor, pero que posteriormente puede generar toxicidad. La megalina participa en la endocitosis y la internalización de la APP121 y de los péptidos Aβ, y se ha descrito en neuronas hipocámpicas un complejo entre megalina-APP y Fe65 que actuaría como regulador en la toxicidad por los péptidos Aβ122. El potencial papel de la APP y su vía de señalización ha sido revisado recientemente por parte de los autores de este artículo123, partiendo del hallazgo de que la mielina fija trazadores de amiloide en PET124. La APP está sobrerregulada en axones desmielinizados y supone la activación de la cascada amiloide favoreciendo la producción de Aβ125,126. La expresión de APP también aumenta tras la lesión compresiva en la sustancia blanca medular en la rata127. VD produce un aumento en la expresión de APP en ratas128 y se ha hipotetizado su papel en la remielinización.
ConclusionesLa capacidad de remielinización se pierde durante las fases avanzadas de la EM81,129. Este aspecto es relevante en el tratamiento de los pacientes con EM, ya que la actual terapéutica solo es efectiva en el control de los mecanismos inmunitarios y en consecuencia en la fases iniciales de la enfermedad, pero no tienen acción en la remielinización y, en consecuencia, en la aparición de secuelas130,131. El metabolismo de la VD está presente en el SNC, participa en el proceso de mielinización y puede estar influido por acciones externas como la dieta, la exposición al sol o la administración de suplementos. El conocimiento de los mecanismos básicos de los efectos de la VD en la mielinización podrá permitir en el futuro poder aconsejar de forma más precisa a los pacientes con EM, cual es la actitud a realizar ante la observación de una deficiencia de VD.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

