




Wernicke's encephalopathy (WE) is an underdiagnosed condition, usually associated with alcoholism, and has a worse prognosis if there is a delay in diagnosis. A series of 8 non-alcoholic patients with WE is presented and an assessment is made on whether a delay in diagnosis leads to a worse prognosis.
Patients and methodsThe clinical records of patients admitted to 2 university hospitals between 2004 and 2009 with the diagnosis of WE, excluding those with a history of alcoholism, were retrospectively reviewed.
ResultsThe study included 4 men and 4 women aged 35–82 of whom 7 had a history of gastrointestinal pathology, and persistent vomiting was the precipitating factor in 7. Encephalopathy was the most frequent onset symptom (4). The classical triad was present in seven patients. Thiamine levels were low in 3/6 and normal in 3/6 cases. MRI was abnormal in seven patients, with high signal intensity in the diencephalon and mammillary bodies (7), periaqueductal grey matter (6), cortex (3) and cerebellum (1). Seven improved with thiamine. Sequelae were mild in 6, and severe in 2 after 6–12 months of follow-up. All patients with a diagnostic delay less than 18 days had mild sequelae.
ConclusionsNon-alcoholic WE frequently occurs after gastrointestinal disturbances that could result in lower thiamine absorption. Whereas thiamine levels can be normal in many cases, in almost all cases the MRI shows signal alterations in typical locations. A delay in the diagnosis, and therefore, in treatment leads to a worse prognosis.
La encefalopatía de Wernicke (EW) es una entidad infradiagnosticada, generalmente asociada a alcoholismo, que tiene peor pronóstico si existe retraso diagnóstico. Se presenta una serie de 8 pacientes no alcohólicos con EW y se evalúa si el retraso en el diagnóstico implica un peor pronóstico.
Pacientes y métodosRevisión retrospectiva de las historias clínicas de pacientes ingresados en dos hospitales universitarios entre 2004 y 2009 con diagnóstico de EW, excluidos aquéllos con historia de alcoholismo.
ResultadosSe incluyó a 4 varones y 4 mujeres, con edades comprendidas entre los 35 y los 82 años; 7 tenían antecedentes patológicos gastrointestinales y los vómitos persistentes fueron el desencadenante en 7 casos. La encefalopatía fue la forma de inicio más frecuente (4 casos). La tríada clásica llegó a estar presente en 7 pacientes. Los niveles de tiamina fueron bajos en 3/6 y normales en 3/6 pacientes. La RM fue patológica en 7 pacientes, con hiperintensidad en diencéfalo y cuerpos mamilares (7), sustancia gris periacueductal (6), corteza (3) y cerebelo (1). Siete pacientes mejoraron tras el tratamiento con tiamina. Las secuelas fueron leves en 5 casos y graves en 3 pacientes. Todos los pacientes con un retraso diagnóstico inferior a 18 días tuvieron secuelas leves.
ConclusionesEn la EW no alcohólica son frecuentes los antecedentes gastrointestinales que podrían condicionar una menor absorción de tiamina. Mientras que los niveles de tiamina pueden ser normales en muchos casos, la RM casi siempre muestra alteración de señal en localizaciones típicas. El retraso en el diagnóstico y, por tanto, en el tratamiento podría implicar un peor pronóstico.
Wernicke's encephalopathy (WE) is an acute neurological syndrome characterised by the classic triad of encephalopathy, ophthalmoplegia and/or nystagmus and ataxia. However, only 16% of patients show all the symptoms at the beginning, so it is considered as an under-diagnosed disorder.1 In autopsy series of patients with WE, it was only diagnosed in life in one third of alcoholic patients and in 6% of non-alcoholics.2 The presence of the classic triad appears to be more common in alcoholics (53.9%) than in non-alcoholics (33.6%).2
Besides the classic triad, patients with WE may also present vestibular dysfunction, peripheral neuropathy, hypothermia, hypotension, syncope, tachycardia, dyspnoea, elevated cardiac rhythm or ECG alterations.3
The diagnosis is clinical and it should be considered in the differential diagnosis of any patient with acute confusion syndrome or acute ataxia. There are no diagnostic laboratory tests and the determination of erythrocyte thiamine transketolase or plasma thiamine levels have technical limitations and low specificity.3 An MRI is useful to support the diagnosis and rule out other causes but should not delay treatment.2,4
The treatment involves immediate thiamine replacement for 3–5 days.5 Using below-therapeutic doses or lack of treatment can lead to death in 20% of these patients or to an irreversible form of anterograde amnesia (Korsakoff's syndrome) in 85% of survivors.4 There are no quality clinical trials that can guide recommendations for a given dose or length of treatment; the EFNS2 recommends administering 200mg intravenously 3 times a day (grade C recommendation) before giving carbohydrates, establishing a normal diet after starting thiamine and maintaining treatment until no further clinical improvement is observed (good clinical practice).
In developed countries, over 80% of WE cases occur in the context of malnutrition associated with alcohol abuse.6 However, WE has also been described in other disorders associated with decreased intestinal absorption of thiamine (low intake or malabsorption), an increase of the body requirements (as in systemic diseases) or loss of soluble thiamine (dialysis). Not all individuals with a similar degree of malnutrition and alcoholism develop WE, so it is believed that there are genetic and environmental factors contributing to the expression of this disease.4
According to a recent review2 of 625 cases reported in the literature, the most frequent causes of WE in non-alcoholic patients were neoplastic disease (18.1%), gastrointestinal surgery (16.8%), hyperemesis gravidarum (12.2%) and fasting or malnutrition (10.2%). Table 1 details the possible causes.
Causes of non-alcoholic WE reported in the literature.
| Gastrointestinal surgery |
| Gastrectomy, gastrojejunostomy, colectomy, bariatric surgery, gastroplasty, intragastric balloon |
| Gastrointestinal alterations |
| Peptic ulcer, ulcerative colitis with megacolon, recurrent vomiting or chronic diarrhoea (caused by pyloric stenosis, gastritis, gallstones, Crohn's disease, intestinal obstruction or perforation, migraine crises, anorexia nervosa, pancreatitis, hyperemesis gravidarum) |
| Cancer and chemotherapy |
| Gastric cancer, colon cancer, non-Hodgkin's lymphoma, myelomonocytic leukaemia, large B-cell lymphoma, myeloid leukaemia, allogeneic bone marrow transplantation, treatment with erbuzole and ifosfamide |
| Systemic diseases |
| Kidney disease with peritoneal dialysis and haemodialysis, AIDS, chronic infections with fever, thyrotoxicosis. Magnesium depletion by diuretics, bowel resection or Crohn's disease |
| Malnutrition |
| Dietary restrictions (psychogenic or due to lack of resources or to treat obesity), negligence due to advanced age or Alzheimer's disease, parenteral nutrition without thiamine supplementation, intravenous hyperalimentation |
Adapted from Sechi and Serra,4 2007.
The relatively simple treatment of this disorder, which can lead to death or chronic neurological deficits if not treated properly, should maintain physicians at high level of clinical suspicion, even when there is no history of alcoholism. In a recent clinical practice guideline, the EFNS recommended suspecting WE in any disorder that can lead to thiamine deficiency and even administering thiamine to any subject at risk who attends the Emergency Room. Moreover, it also recommended thiamine supplementation in patients undergoing bariatric surgery.
The aim of this study was to analyse a series of 8 non-alcoholic patients who developed WE, as well as to evaluate whether a delay in diagnosis may have influenced their prognosis.
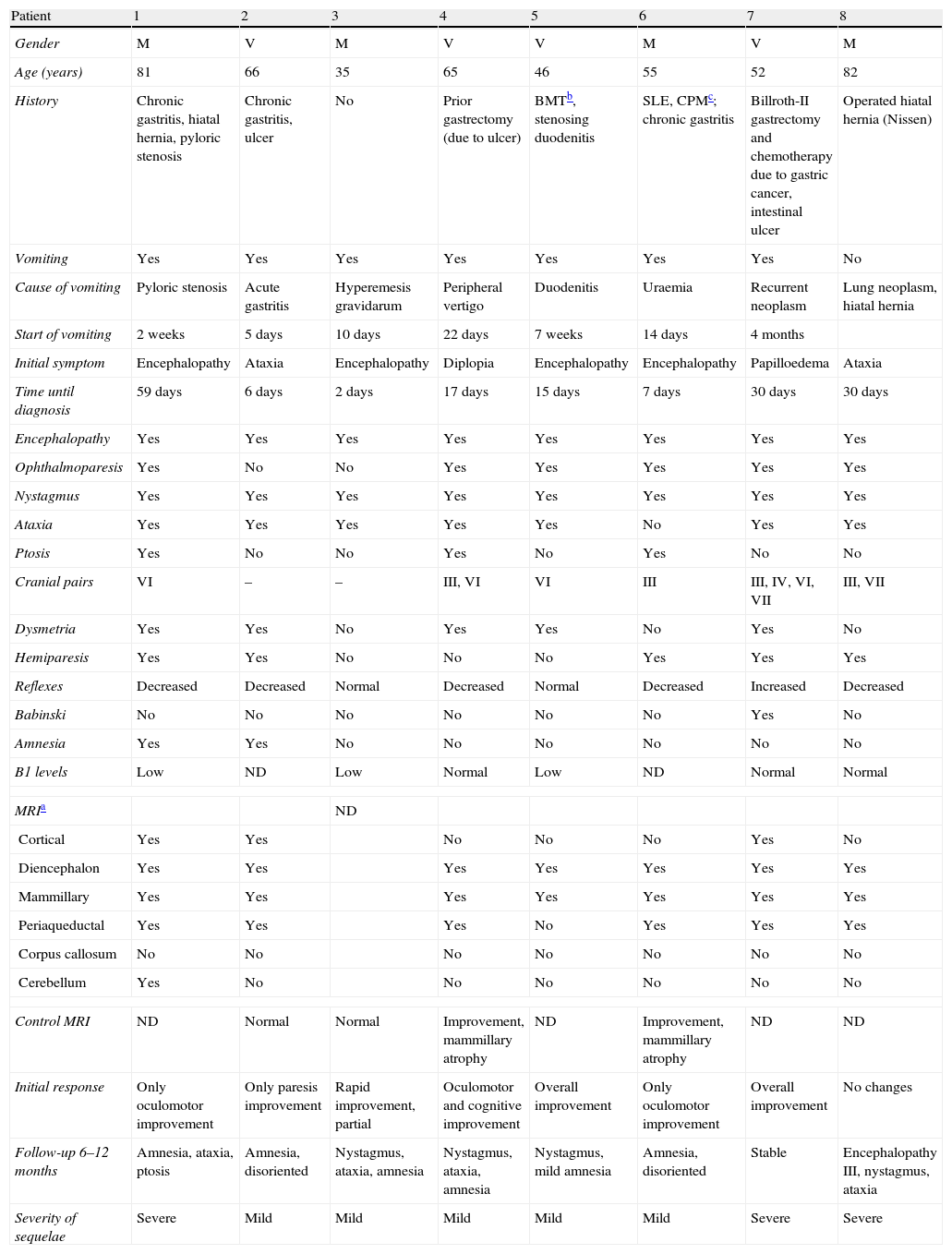
Patients and methodsWe retrospectively reviewed databases including the medical records of patients admitted to two university hospitals between 2004 and 2009, seeking the diagnosis of WE. Of these, we considered those who presented at least 2 of the 3 signs of the classic triad: encephalopathy, ophthalmoplegia and/or nystagmus and ataxia. Patients with a history of alcoholism were excluded. Finally, we included 8 patients (4 men and 4 women). We recorded their medical histories, especially gastrointestinal antecedents, presentation symptoms, neurological findings, delay between symptom onset and diagnosis, cranial MRI findings, plasma levels of thiamine, response to treatment and presence of residual deficits. Patients were considered as with no sequelae or mild sequelae when they had a score ≤3 on the Modified Rankin Scale7 and with a normal cognitive status or mild cognitive impairment (according to the Petersen8 criteria), and with severe sequelae when they presented a score above 3 or suffered a greater cognitive impairment than mild. These data are summarised in Table 2.
Clinical and neuroimaging data from non-alcoholic patients with WE.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Gender | M | V | M | V | V | M | V | M |
| Age (years) | 81 | 66 | 35 | 65 | 46 | 55 | 52 | 82 |
| History | Chronic gastritis, hiatal hernia, pyloric stenosis | Chronic gastritis, ulcer | No | Prior gastrectomy (due to ulcer) | BMTb, stenosing duodenitis | SLE, CPMc; chronic gastritis | Billroth-II gastrectomy and chemotherapy due to gastric cancer, intestinal ulcer | Operated hiatal hernia (Nissen) |
| Vomiting | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Cause of vomiting | Pyloric stenosis | Acute gastritis | Hyperemesis gravidarum | Peripheral vertigo | Duodenitis | Uraemia | Recurrent neoplasm | Lung neoplasm, hiatal hernia |
| Start of vomiting | 2 weeks | 5 days | 10 days | 22 days | 7 weeks | 14 days | 4 months | |
| Initial symptom | Encephalopathy | Ataxia | Encephalopathy | Diplopia | Encephalopathy | Encephalopathy | Papilloedema | Ataxia |
| Time until diagnosis | 59 days | 6 days | 2 days | 17 days | 15 days | 7 days | 30 days | 30 days |
| Encephalopathy | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Ophthalmoparesis | Yes | No | No | Yes | Yes | Yes | Yes | Yes |
| Nystagmus | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Ataxia | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes |
| Ptosis | Yes | No | No | Yes | No | Yes | No | No |
| Cranial pairs | VI | – | – | III, VI | VI | III | III, IV, VI, VII | III, VII |
| Dysmetria | Yes | Yes | No | Yes | Yes | No | Yes | No |
| Hemiparesis | Yes | Yes | No | No | No | Yes | Yes | Yes |
| Reflexes | Decreased | Decreased | Normal | Decreased | Normal | Decreased | Increased | Decreased |
| Babinski | No | No | No | No | No | No | Yes | No |
| Amnesia | Yes | Yes | No | No | No | No | No | No |
| B1 levels | Low | ND | Low | Normal | Low | ND | Normal | Normal |
| MRIa | ND | |||||||
| Cortical | Yes | Yes | No | No | No | Yes | No | |
| Diencephalon | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Mammillary | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Periaqueductal | Yes | Yes | Yes | No | Yes | Yes | Yes | |
| Corpus callosum | No | No | No | No | No | No | No | |
| Cerebellum | Yes | No | No | No | No | No | No | |
| Control MRI | ND | Normal | Normal | Improvement, mammillary atrophy | ND | Improvement, mammillary atrophy | ND | ND |
| Initial response | Only oculomotor improvement | Only paresis improvement | Rapid improvement, partial | Oculomotor and cognitive improvement | Overall improvement | Only oculomotor improvement | Overall improvement | No changes |
| Follow-up 6–12 months | Amnesia, ataxia, ptosis | Amnesia, disoriented | Nystagmus, ataxia, amnesia | Nystagmus, ataxia, amnesia | Nystagmus, mild amnesia | Amnesia, disoriented | Stable | Encephalopathy III, nystagmus, ataxia |
| Severity of sequelae | Severe | Mild | Mild | Mild | Mild | Mild | Severe | Severe |
ND: not done.
The patient was an 81-year-old woman with a history of hiatal hernia, chronic gastritis and pyloric stenosis. She was admitted due to subacute cognitive impairment with memory deficit, disorientation and apraxia after vomiting for 2 weeks, which was attributed to pyloric stenosis. Upon admission, she presented a limitation of the upward gaze and bilateral abduction, horizontal and vertical nystagmus, and ataxia. She also presented progressive bilateral ptosis, decreased bilateral visual acuity, distal hypoesthesia, anterograde amnesia, mild right dysmetria, mild right hemiparesis and areflexia of the lower limbs. Due to the presence of the classic triad, WE was suspected. Thiamine levels were measured and appeared to be low, so thiamine was administered. The time between the onset of clinical symptoms and diagnosis was 59 days. The MRI scan showed signal alterations in cortical regions, diencephalon, mammillary bodies, periaqueductal area and cerebellum. Pyloric stenosis improved with Helicobacter pylori eradication therapy. The initial response to thiamine was good, with improvement of oculomotor deficits and hemiparesis; however, disorientation, ataxia and amnesia lingered. At 6 months, the patient had improved in the oculomotor deficits and PNP, but still presented memory loss, disorientation, ataxia and bilateral ptosis as sequelae.
Patient 2The patient was a 66-year-old male with a history of chronic obstructive pulmonary disease, peripheral vertigo, gastric ulcer and chronic gastritis. He suffered vomiting secondary to acute gastroenteritis during 5 days and subsequently developed gait ataxia and was admitted to hospital. Exploration found anterograde amnesia, disorientation and horizontal nystagmus in all directions (but without ophthalmoplegia), clumsiness of the right hand, left dysmetria and mild hemiparesis, hypopallesthesia and areflexia in the lower limbs. He was given thiamine due to suspicion of WE, but levels were not determined. The time from the onset of neurological symptoms to diagnosis was 6 days. An MRI scan showed signal alterations in cortical, diencephalic, periaqueductal and mammillary regions. Treatment with thiamine resolved only the hemiparesis, while ataxia, nystagmus and amnesia persisted. A subsequent control MRI performed 1 month after treatment was normal. Sequelae at 6 months were anterograde amnesia and time-space disorientation.
Patient 3The patient was a 35-year-old female who presented vomiting due to hyperemesis gravidarum for 10 days in the ninth month of pregnancy, after which she developed a subacute case of cognitive impairment and bradypsychia compatible with encephalopathy, leading to hospital admission. Examination revealed ataxia, dysarthria and vertical nystagmus without ophthalmoparesis. The time to diagnosis was only 2 days. Thiamine levels were low. No MRI scan was performed at the start and the control MRI performed 15 days after treatment was normal. The initial response was good, with improvement of encephalopathy and dysarthria, but the patient presented anterograde amnesia, nystagmus and ataxia as sequelae at 6 months.
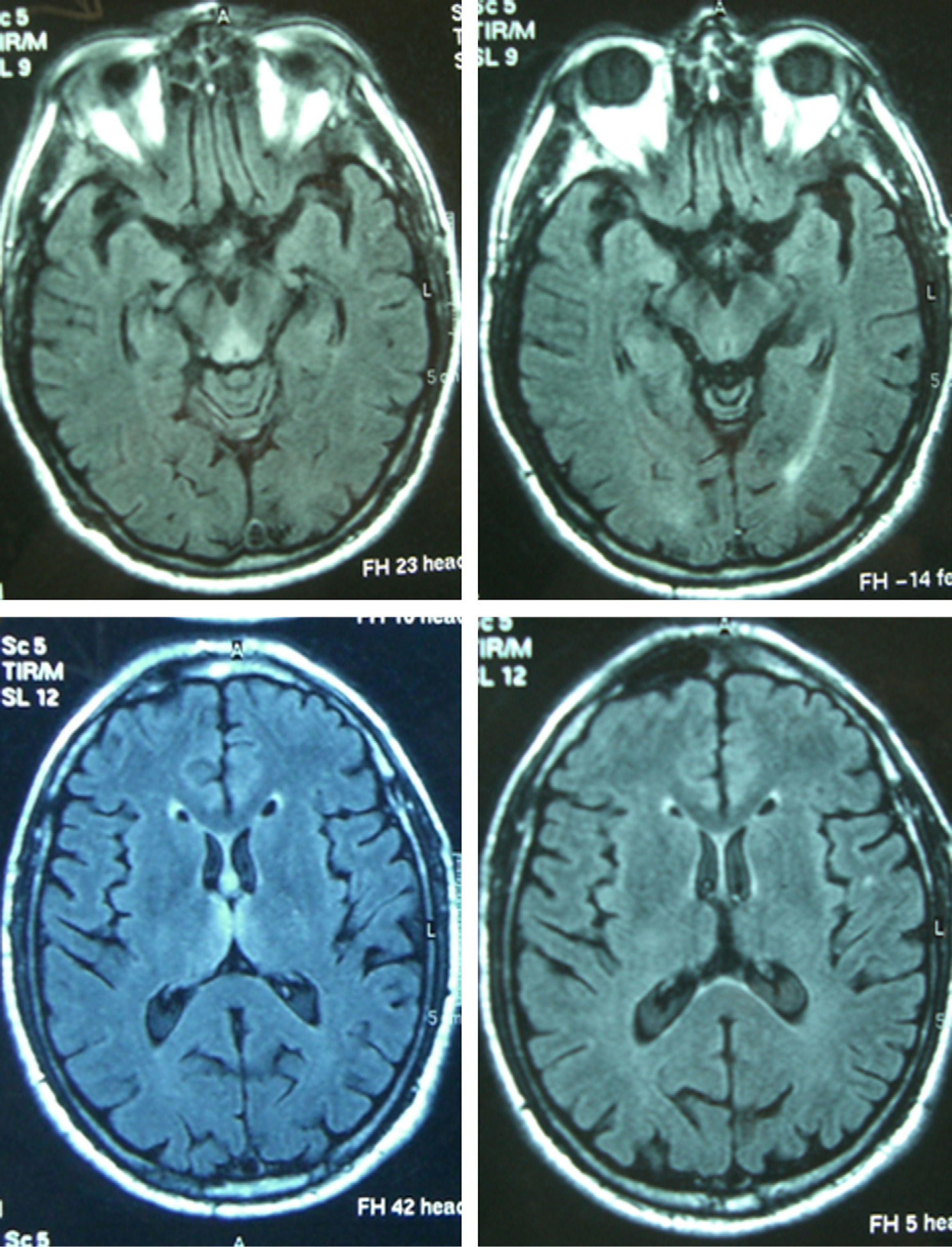
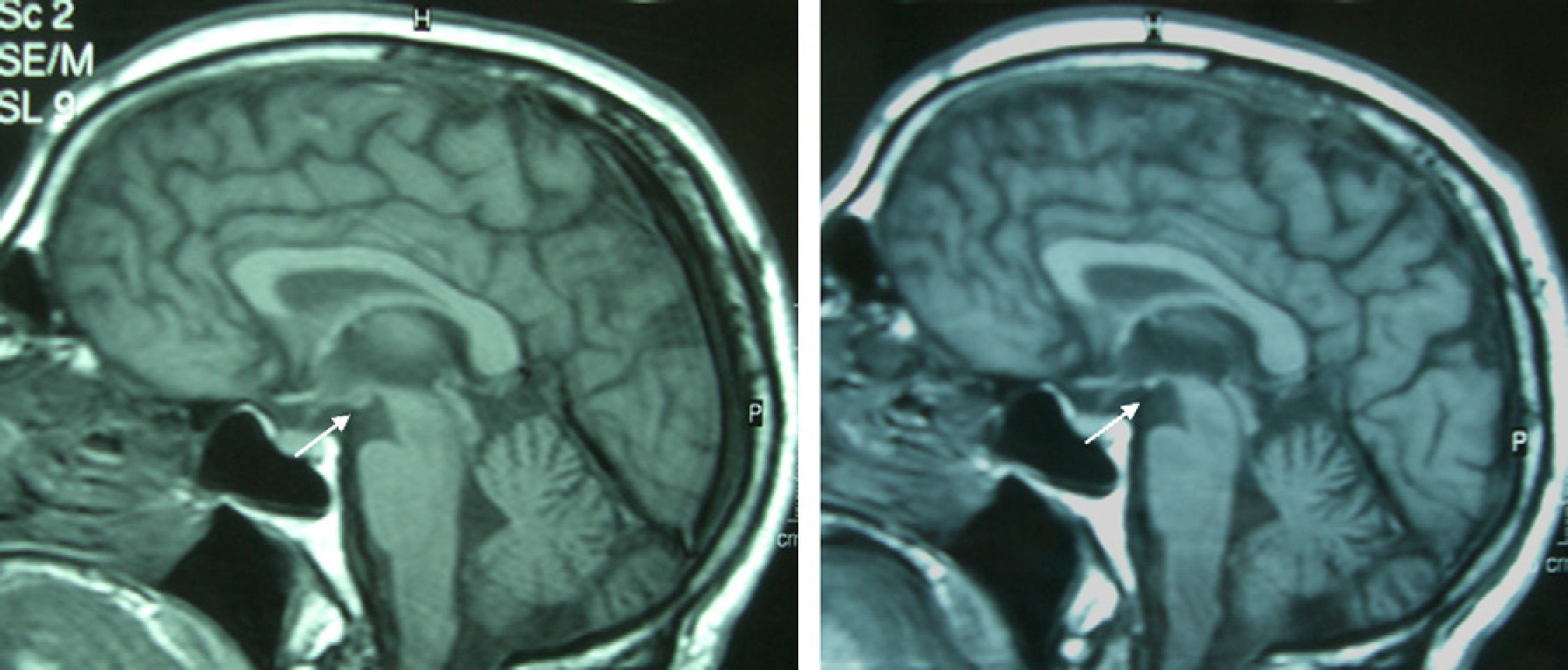
Patient 4The patient was a 65-year-old male with a history of gastrectomy due to gastroduodenal ulcer. He suffered vomiting due to peripheral vertigo for 22 days and then presented diplopia and bilateral ptosis. Examination on admission found the complete triad with encephalopathy, sixth nerve paresis, paralysis of downwards and right gaze, vertical nystagmus and ataxia, as well as bilateral ptosis and dysmetria and areflexia in lower limbs. The time from the onset of clinical symptoms until diagnosis was 17 days. Thiamine levels were normal. The MRI showed a signal alteration in the diencephalon, mammillary bodies and periaqueductal area (Fig. 1). The initial response to thiamine was good, with improvement of encephalopathy and ophthalmoparesis. A control MRI performed 9 months after treatment showed an improvement of signal alteration in the thalamus, hypothalamus and periaqueductal area, and atrophy of the mammillary bodies (Fig. 2). Amnesia, vertical nystagmus and unsteady tandem gait persisted after 9 months follow-up.
 signals, with clear improvement in the control MRI (right).")
.")
The patient was a 46-year-old male with arterial hypertension, peripheral vascular disease and acute myeloid leukaemia, who was treated with bone marrow transplantation, achieving complete remission. Three months after transplantation, he suffered vomiting due to non-erosive duodenitis, which persisted for 7 weeks. After that, he began to present clinical symptoms of subacute cognitive impairment with disorientation, amnesia and bradypsychia compatible with encephalopathy. Subsequently, the other symptoms of the clinical triad appeared during admission: bilateral sixth nerve palsy, nystagmus and ataxic gait, as well as right dysmetria. The time from the onset of neurological symptoms to diagnosis was 15 days. Thiamine levels were low. An MRI scan showed hyperintense signal in the diencephalon and mammillary bodies. The initial response to treatment was good, with improvement of all clinical signs. At 6 months, he still suffered a slight memory loss and nystagmus.
Patient 6The patient was a 55-year-old woman with arterial hypertension, atrial fibrillation, depression, migraine, chronic liver disease from hepatitis C virus and lupus nephritis. She followed treatment with corticosteroids and cyclophosphamide. She was admitted due to vomiting of 14 days’ duration, associated with worsening of renal function. Symptoms remitted after the correction of uraemia. Subsequently, she presented encephalopathy, which was associated with bilateral third nerve palsy and horizontal nystagmus, but no ataxia. She also developed right ptosis, generalised weakness and hyporeflexia. Axonal PNP was demonstrated by electromyography. An upper oesophago-gastroduodenal examination revealed a lack of oesophageal peristalsis and a gastroscopy with biopsy found focally active chronic gastritis. Thiamine levels were not determined. The MRI scan showed an altered signal in the diencephalon, mammillary bodies and periaqueductal area. The time to diagnosis was 7 days. The initial response to thiamine was good, with normalisation of ocular motility. A control MRI performed after 6 months of treatment showed great improvement and residual atrophy of the mammillary bodies. Nevertheless, deficits of recent memory and disorientation persisted as sequelae.
Patient 7The patient was a 52-year-old male with a history of gastric cancer treated with gastrectomy and chemotherapy 8 years earlier. He suffered vomiting for 4 months due to tumour recurrence with intestinal ulcer as well as oral intolerance, which required admission and parenteral nutrition. During admission, he began to experience loss of vision. An ophthalmologic examination revealed bilateral papilloedema and achromatopsia. Subsequently, the classic triad of symptoms appeared, with bilateral paralysis of the third, fourth and sixth cranial nerves, nystagmus in all directions, encephalopathy and ataxia. In addition, the patient suffered left facial palsy, mild hemiparesis, left dysmetria, right Babinski sign and hyperreflexia. The time from the onset of neurological symptoms to diagnosis was 30 days. Thiamine levels were normal. An MRI scan showed hyperintense cortical regions in the diencephalon, the mammillary bodies and periaqueductal area. The initial response to thiamine was good, with resolution of most symptoms; however, mild nystagmus and moderate encephalopathy persisted, still present at 6 months.
Patient 8The patient was an 82-year-old male with hypertension, chronic heart failure and hiatal hernia, who was admitted due to symptoms of gait instability and dyspnoea. Arterial gasometry showed hypoxemia and a chest computed tomography revealed multiple bilateral pulmonary thromboembolisms, as well as the presence of a non-oat-cell lung cancer with multiple lymphadenopathies and a giant hiatal hernia. Subsequently, the classic triad of symptoms appeared, with encephalopathy, bilateral third nerve palsy, horizontal nystagmus and ataxia. The patient also presented mild paresis of all 4 limbs, left facial paralysis and anarthria. An MRI scan showed an altered signal in the diencephalon, mammillary bodies and periaqueductal area. Thiamine levels were normal. The time from the onset of clinical symptoms until diagnosis was 30 days. There was no prior vomiting in this case. Treatment with thiamine did not produce clinical changes.
ResultsThe study included 8 non-alcoholic patients with WE (4 men and 4 women), with an age range between 35 and 82 years. Of these, 7 presented a medical history of gastrointestinal pathology and 3 had a history of current neoplastic processes. Persisting vomiting was the trigger in all except 1 case. The initial symptom was encephalopathy in 4 patients, ataxia in 2, diplopia in 1 and papilloedema in another. The classic triad of encephalopathy, oculomotor alterations and ataxia appeared in all patients except in 1, who did not present ataxia. The most commonly affected cranial nerve was the third (4 patients) and all patients suffered nystagmus. Other common neurological signs were dysmetria (5), hemiparesis (5), hypopallesthesia (2) and impaired reflexes (5 with areflexia, 1 with hyperreflexia).
Thiamine levels were determined in 6 of the 8 patients. They were low in 3 of them and normal in the other 3. The initial MRI was abnormal in all 7 cases in which it was performed, showing hyperintensity on T2 and FLAIR sequences, although hypointensity in T1 was also detected in 3 patients and gadolinium-enhanced lesions were found in 2 patients. The most affected regions were the diencephalon and the mammillary bodies (7), followed by the periaqueductal grey matter (6), cortex (3) and cerebellum (1). A control MRI was performed on 4 patients. This showed residual atrophy of the mammillary bodies in 2 cases (with improvement of the remaining lesions in 1 and persistence in the other) and was normal in the other 2 cases (resolution of lesions in 1 case, while the other had not undergone an initial MRI scan).
Thiamine treatment was administered to all patients, with partial clinical improvement in 7 of them. At 6–12 months follow-up, 5 of them presented mild sequelae and 3 presented severe sequelae. The 3 patients with severe sequelae had suffered a diagnostic delay of 18 or more days, while patients with mild sequelae had suffered a diagnostic delay of less than 18 days.
DiscussionWernicke's encephalopathy is due to thiamine deficiency. Thiamine, in its biologically active form (pyrophosphate), is an essential coenzyme in the metabolism of glucose in the brain.9 The mechanism through which its deficit causes brain lesions is unknown, although it is believed that neuronal damage begins once the metabolism in brain regions with high metabolic requirements and high thiamine turnover is inhibited.
It is important to note that most WE patients do not present the classic triad1,2 and that thiamine levels can be normal in some patients, whereas the MRI scan almost always shows characteristic alterations. Therefore, other causes for WE should be suspected when faced with a patient suffering acute confusional syndrome or acute or subacute ataxia or oculomotor alteration, with or without other neurological symptoms, especially in the context of vomiting, gastrointestinal or neoplastic history. The EFNS2 recommends determining thiamine levels just before starting treatment (good clinical practice) and performing an MRI scan to support the diagnosis (grade B recommendation). According to a clinical-pathological study in alcoholic patients with WE, clinical diagnosis requires the presence of at least 2 of the following signs: nutritional deficiencies, ocular disorders, cerebellar dysfunction and altered mental status or mild memory impairment (grade B recommendation), which achieves a diagnostic sensitivity of 85%.2 The EFNS considers applying the same criteria to non-alcoholic patients reasonable (good clinical practice). All these signs were present in the patients in this series, except in 1 who had no cerebellar signs, which is consistent with the recommendation. Although nutritional deficit is not as evident in non-alcoholic patients, perhaps special attention should be paid to a history of vomiting.
Reversible cytotoxic oedema seems to be the most distinctive lesion in WE and the most useful sequences to detect it are T2, FLAIR and DWI.10,11 The typical lesions are symmetrical and can be observed in the thalamus, mammillary bodies, tectal plate and periaqueductal area. Atypical lesions can be observed in the cerebellum; cerebellar vermis; cranial nerve nuclei: red, dentate, caudate nuclei; splenium; and cerebral cortex.11 The atypical locations have been described mostly in non-alcoholic patients and associated with typical alterations, while gadolinium uptake in the thalamus and mammillary bodies is more common in alcoholics. In the patients of this series, the most affected regions were the typical locations of diencephalon and mammillary bodies (7) and the periaqueductal grey matter (6), followed by the atypical locations of cortex (3) and cerebellum (1). In the latter, the atypical alterations were also associated with alteration in the 3 typical locations. The MRI appears to be more sensitive in detecting WE lesions in non-alcoholic than in alcoholic patients. In a retrospective study in alcoholic patients, the sensitivity and specificity results obtained were 53% and 93%, with a positive predictive value of 89%.12 Pooled data from several studies show that conventional MRI was able to detect changes in two-thirds of the alcoholic subjects with WE. The FL2AIR and DWI images did not provide further information. In contrast, in non-alcoholic patients, MRI detected lesions in up to 97% in DWI, 99% in conventional MRI and 100% in FLAIR.2 The alteration of the T2 signal disappears within 48h after administration of thiamine.13
Atrophy of the mammillary bodies is a very specific pathological finding in chronic WE and Korsakoff's syndrome and is present in 80% of alcoholics with a history of WE.14,15 It can be detected just 1 week after the start of WE.13 In contrast, atrophy of the mammillary bodies has not been found in non-alcoholic WE patients, since it is an acute deficit.11 Although it is not described in the literature, probably due to the small number of patients, it is noteworthy that 2 of the patients in this series presented atrophy of the mammillary bodies in the control MRI performed a few days after treatment; in contrast, it was not present in the initial MRI. This suggests that atrophy of the mammillary bodies is not a finding unique to alcoholic WE and that it can develop in a short time.
It is common for residual deficits to persist in alcoholic patients with WE.3 Oculomotor alterations usually improve in a few hours after administering thiamine, but nystagmus persists in 60% of cases. Only 40% of patients recover from ataxia and over half of patients are left with residual gait disorder. Acute encephalopathy is resolved slowly, but learning deficit and recent memory remain (Korsakoff's psychosis), which are only resolved in 20%.
Except for 1 case, the cause of WE was persistent vomiting due to different causes, mainly gastrointestinal in all patients in this series. The mechanism must have been a decrease in the absorption of thiamine. In the absence of thiamine, the body reserve is depleted within 2–3 weeks, affecting the most vulnerable brain regions, and the plasma levels decrease after 3 weeks.4 However, this reserve can be particularly low in some individuals or be consumed quickly, as in the case of Patient 2, who developed a case of WE after only 5 days of vomiting.
It is likely that some people who present a genetically lower transketolase activity have a higher thiamine requirement and are therefore more predisposed to suffer WE in situations of higher demand or lower absorption. A higher genetic predisposition has been described in identical twins,9 as well as an altered affinity of the transketolase enzyme for thiamine.16 Phenotypic variants have also been detected in the thiamine transporter gene.17 It is essential to start treatment as soon as possible, since a delay may lead to more severe sequelae or further irreversible damage. This is reflected in the fact that the 3 patients with severe sequelae were those who had a diagnostic delay of 18 or more days, while patients with mild sequelae were those diagnosed within 18 days. Thiamine treatment was administered at the time WE was suspected. The delay in diagnosis was due to the fact that, at the onset, patients usually present only 1 or 2 WE symptoms, which in addition are non-specific and could be related to the underlying disease. Thus, WE is often not suspected until the classic triad appears or a suggestive image is identified in MRI. Therefore, we must insist on increasing the level of suspicion in patients at risk.
In conclusion, in alcoholic patients with WE, it is common to find the presence of a gastrointestinal history that could condition lower absorption of thiamine. This would be a physiopathogenic factor in patients with genetic susceptibility. While the levels of thiamine may be normal in many cases, the MRI almost always shows signal alterations in typical locations, thus being more effective for the diagnosis. A delay in diagnosis and, therefore, in treatment can worsen the prognosis.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Gascón-Bayarri J, et al. Encefalopatía de Wernicke en pacientes no alcohólicos: una serie de 8 casos. Neurología. 2011;26:540–7.
