



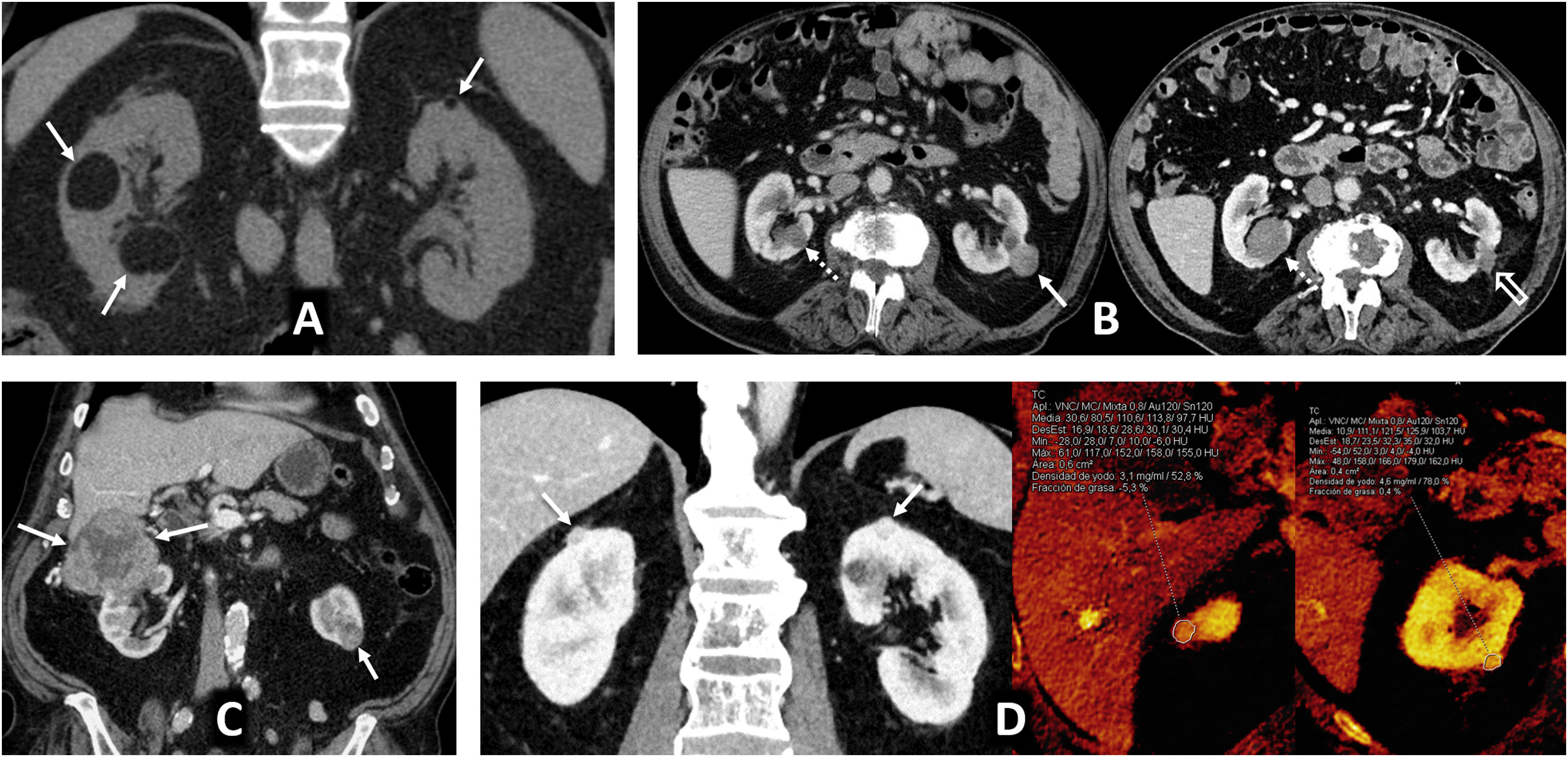
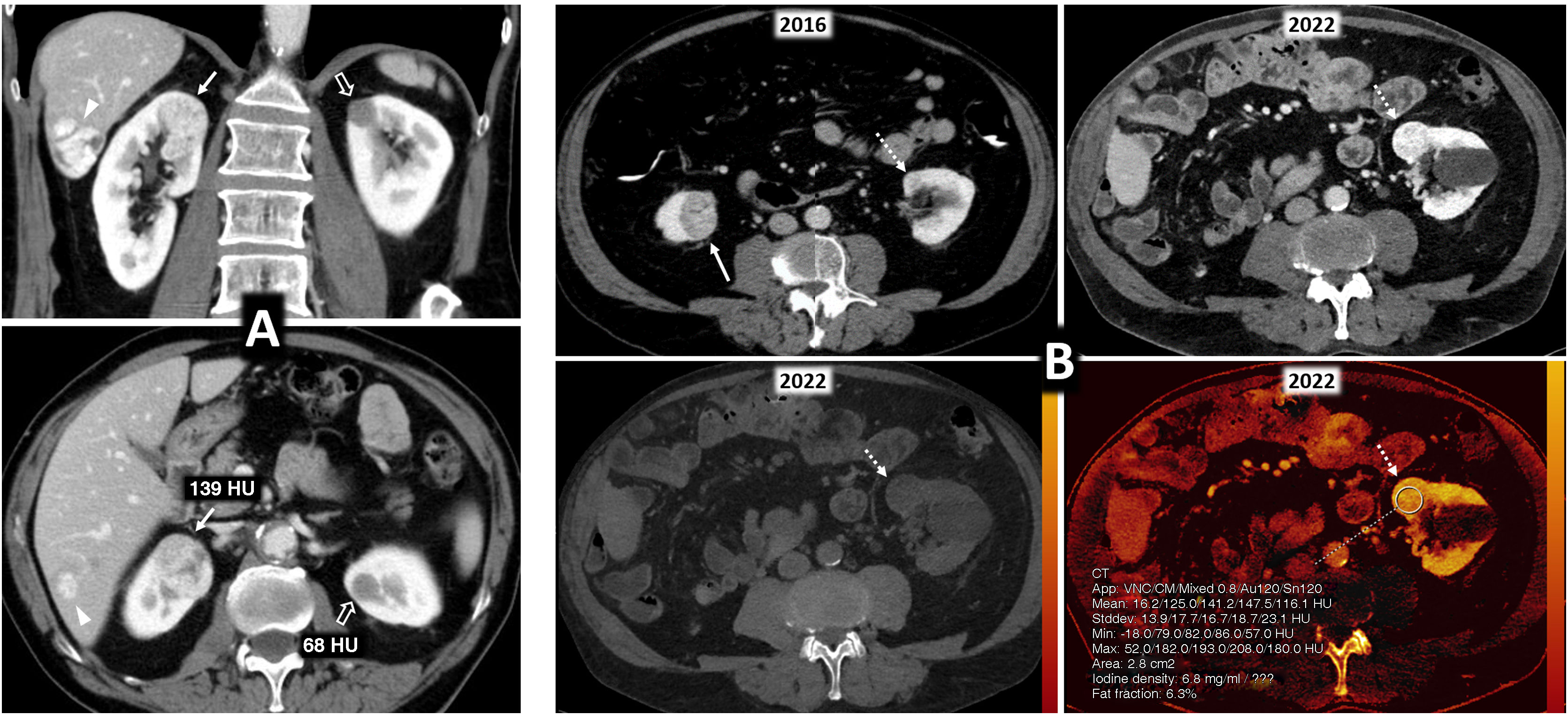
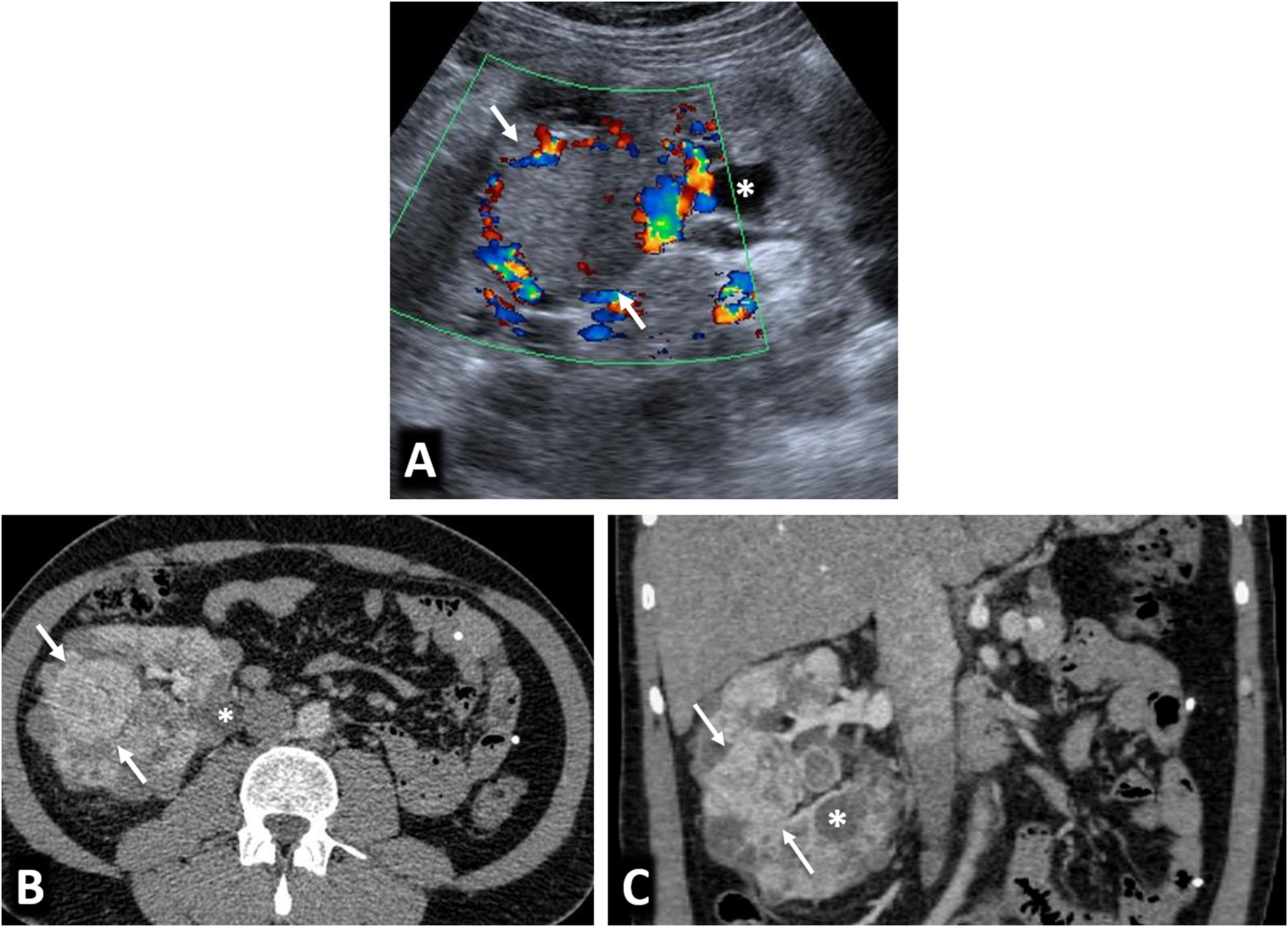
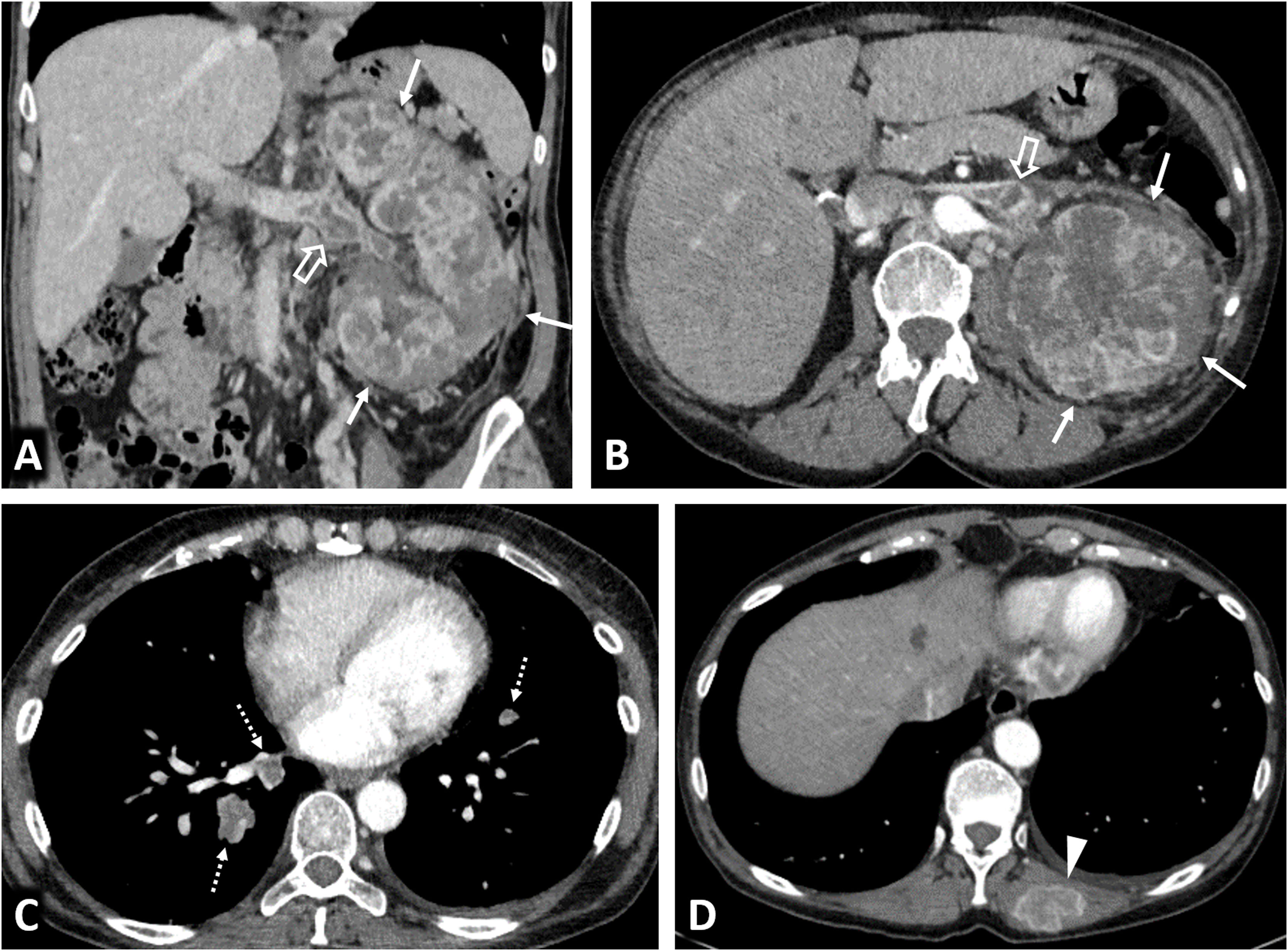
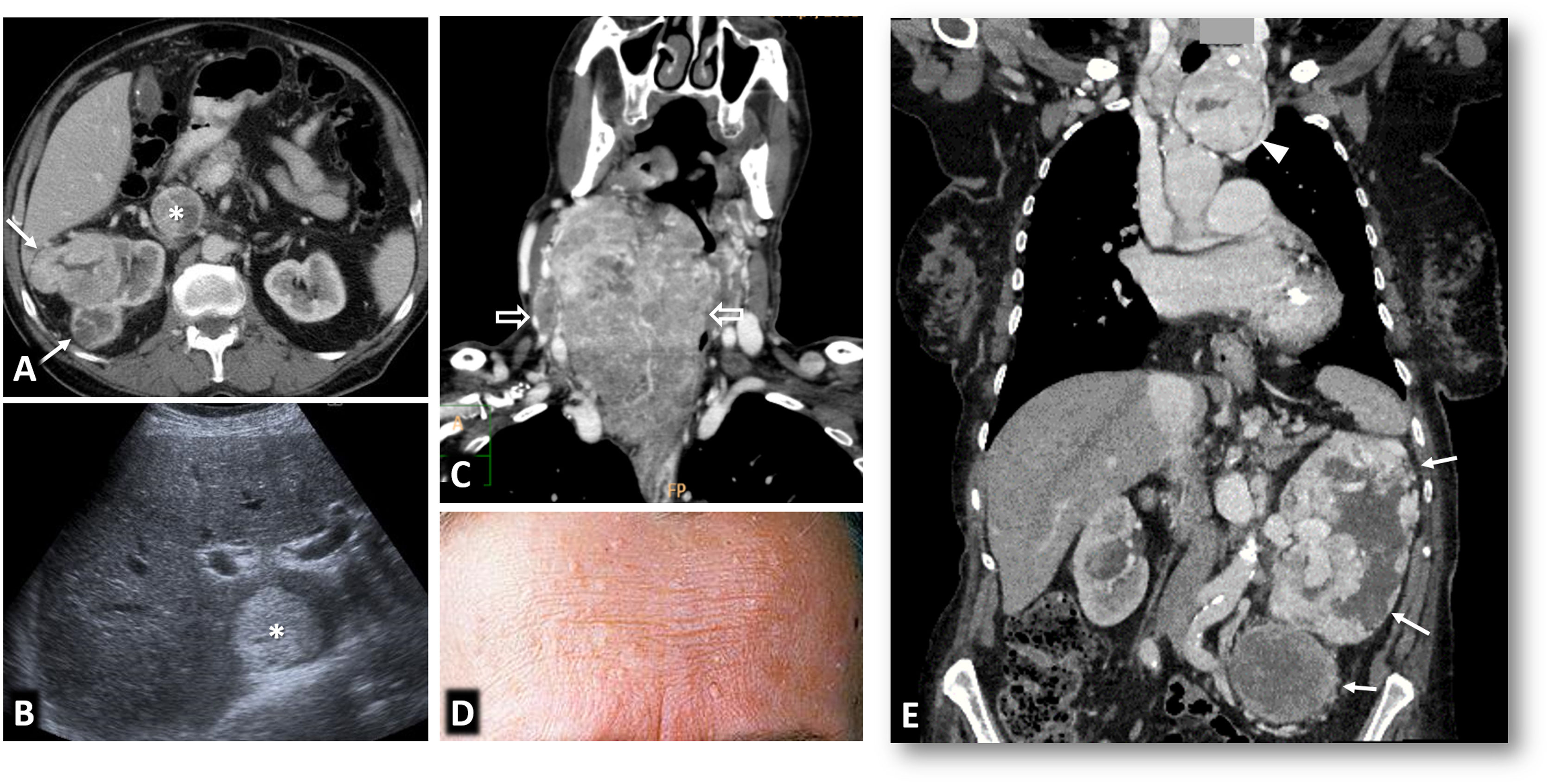
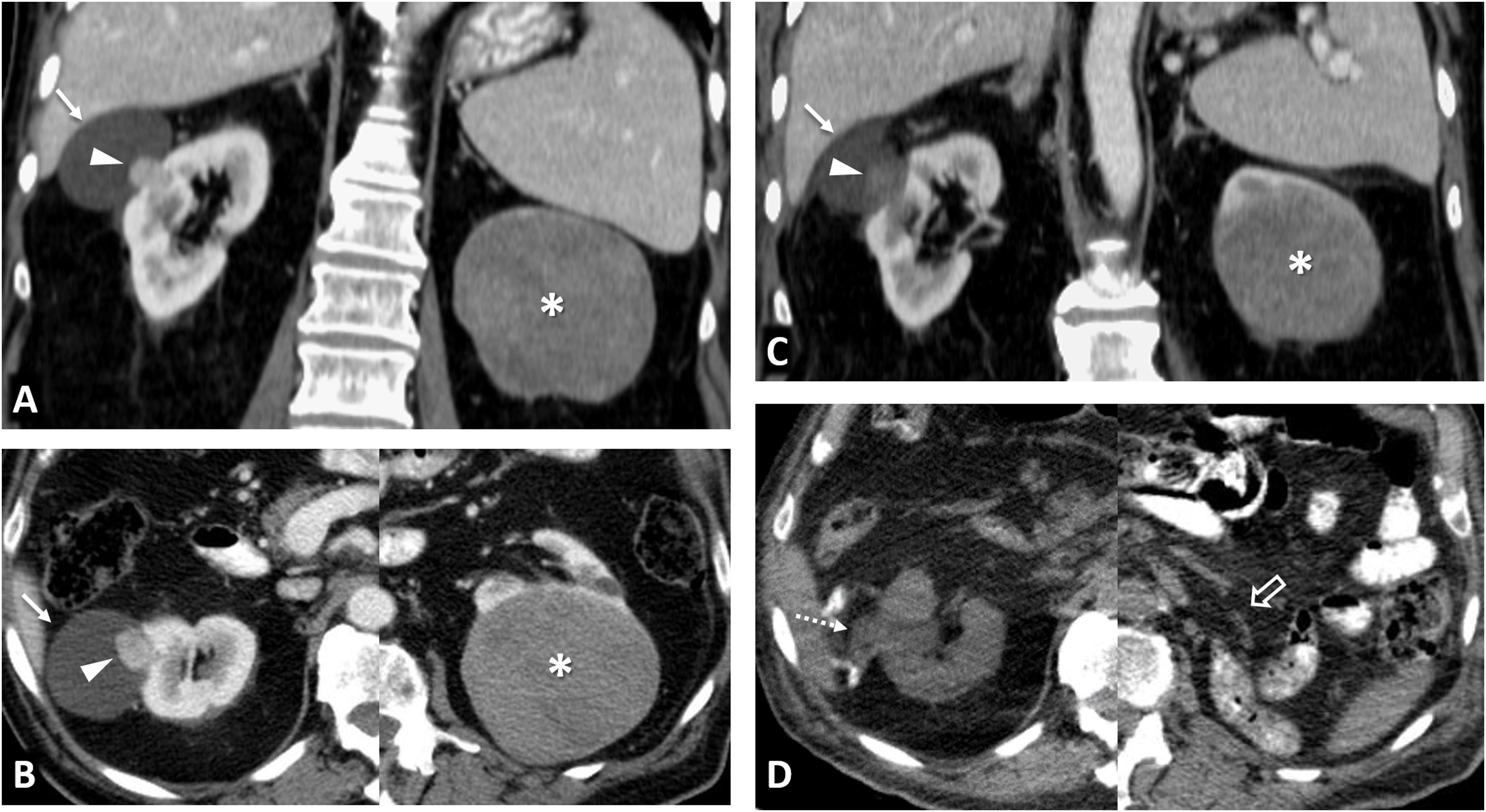
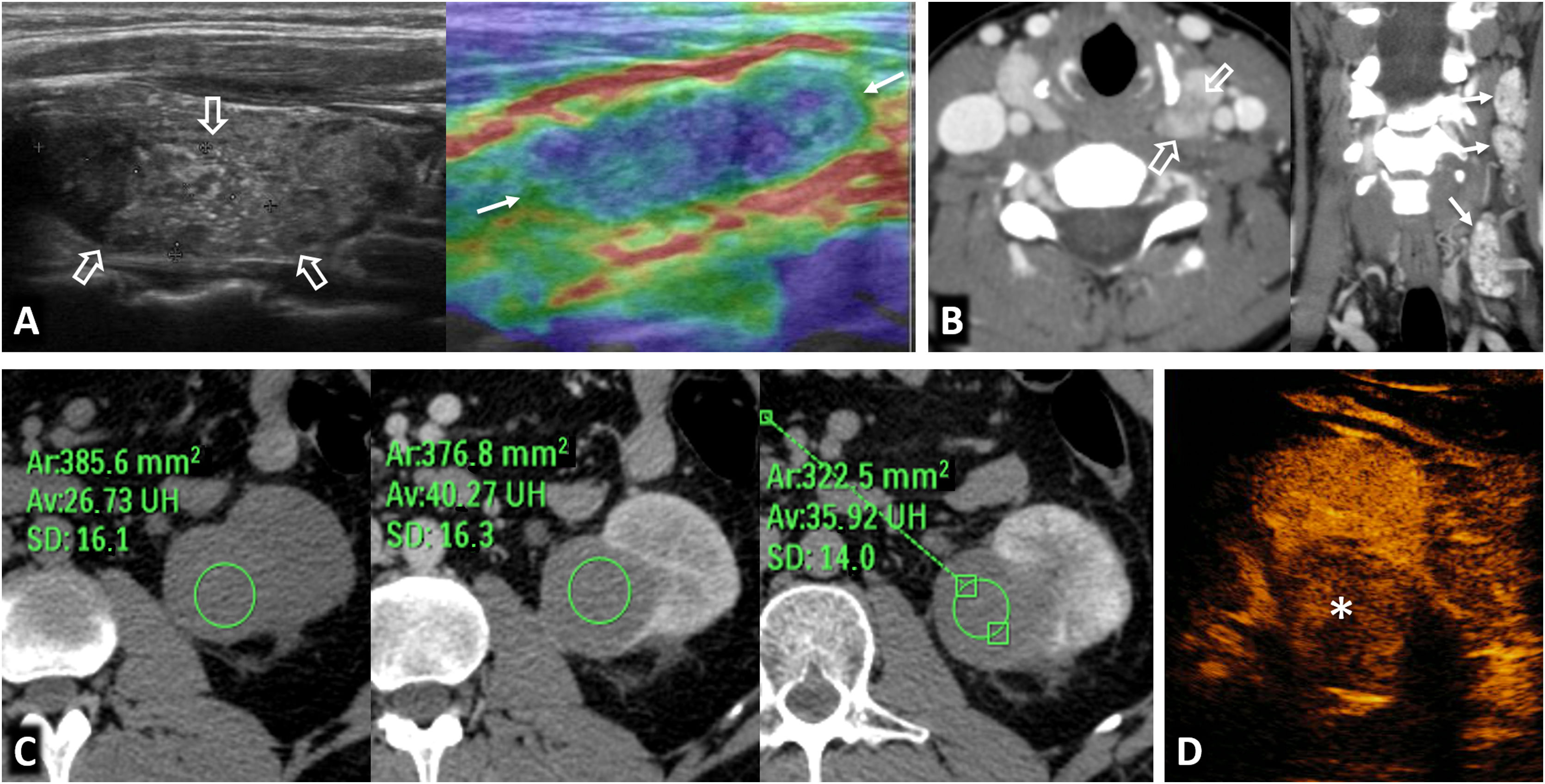
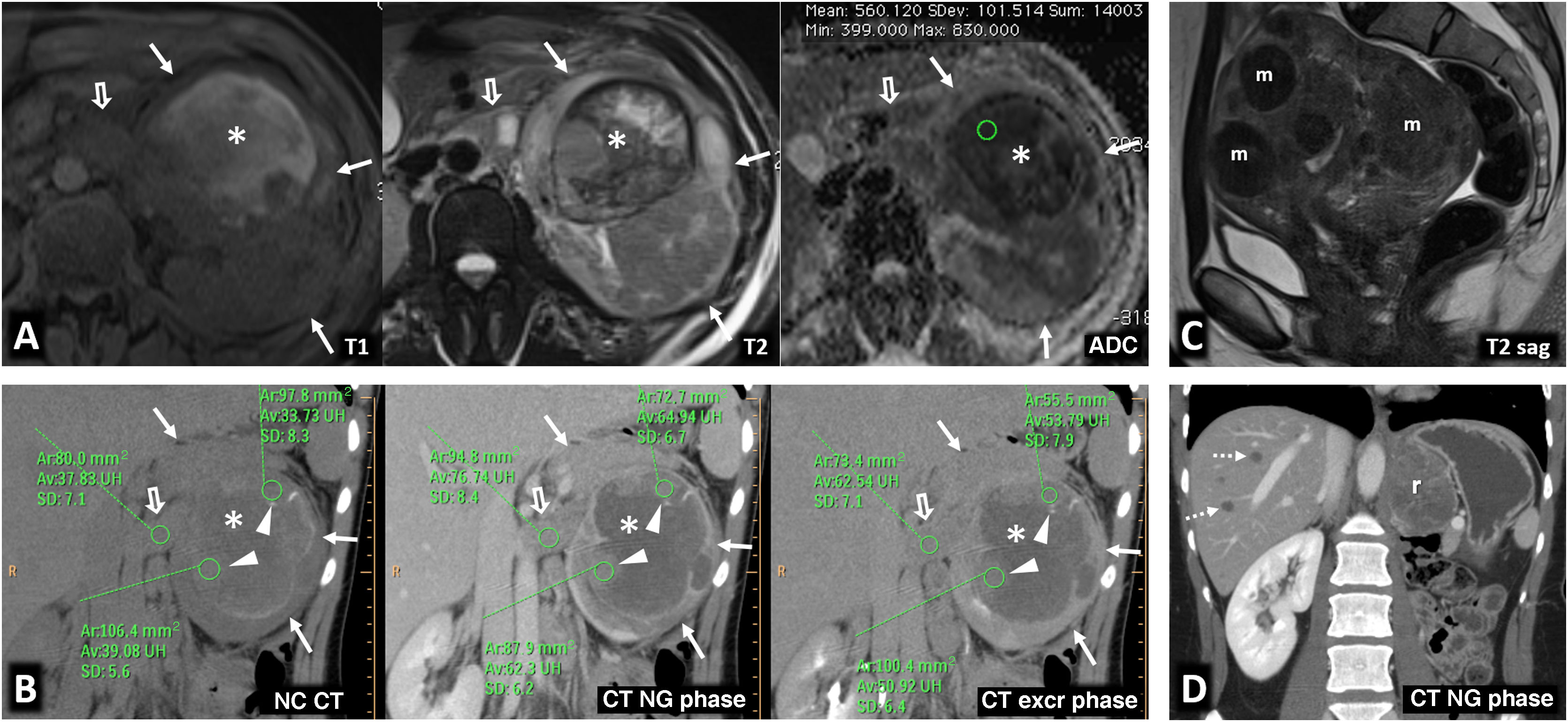
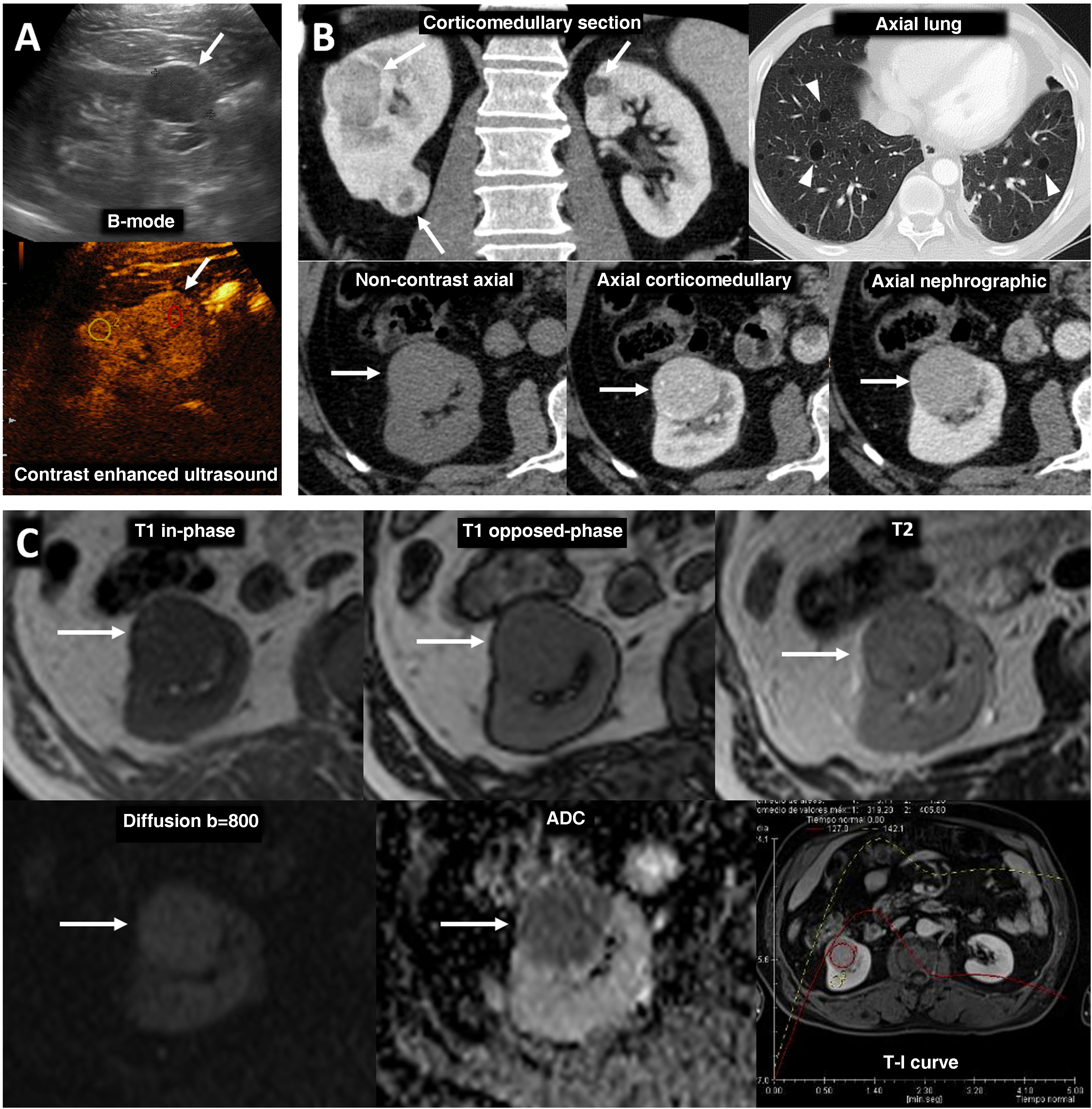
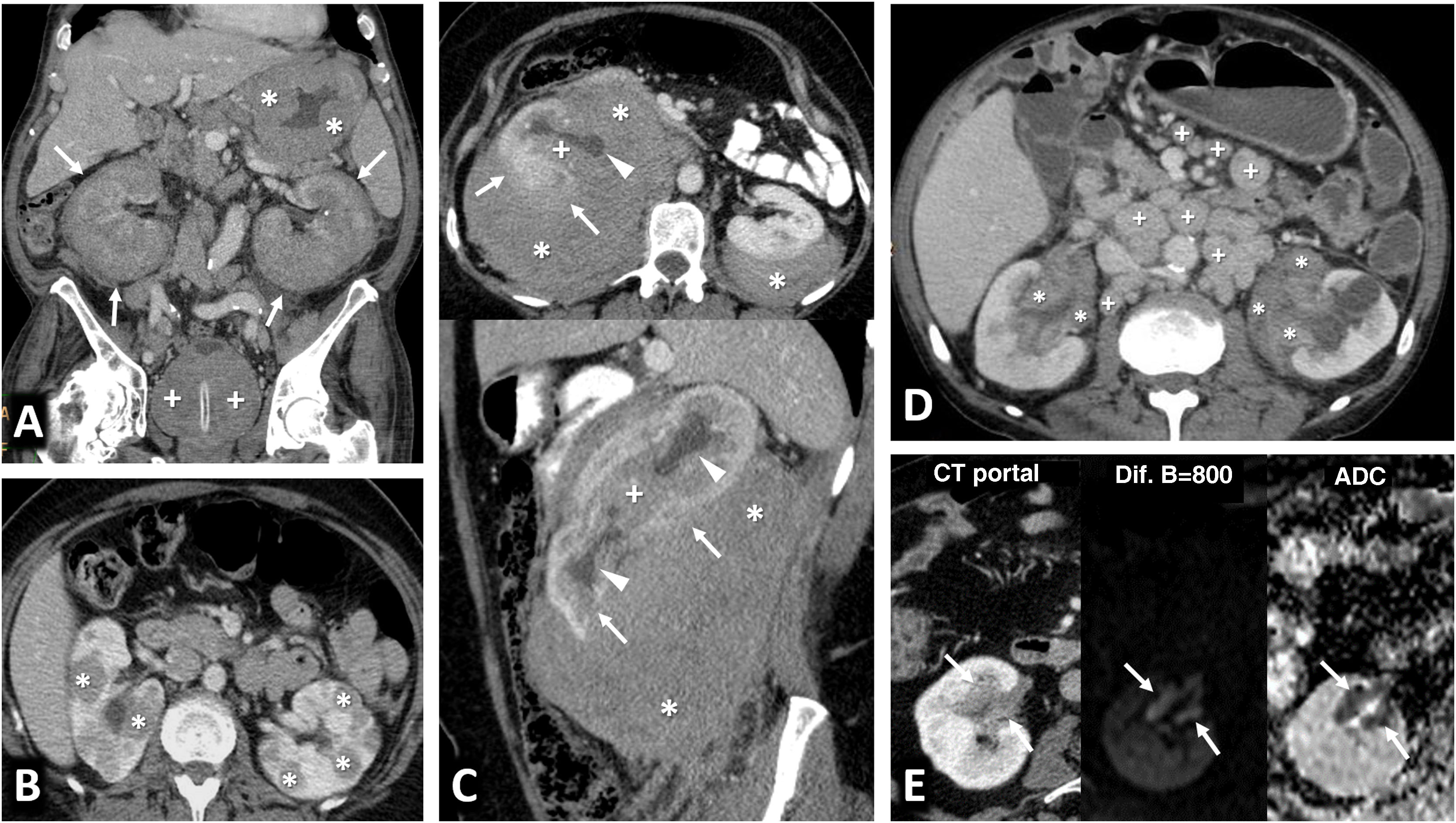
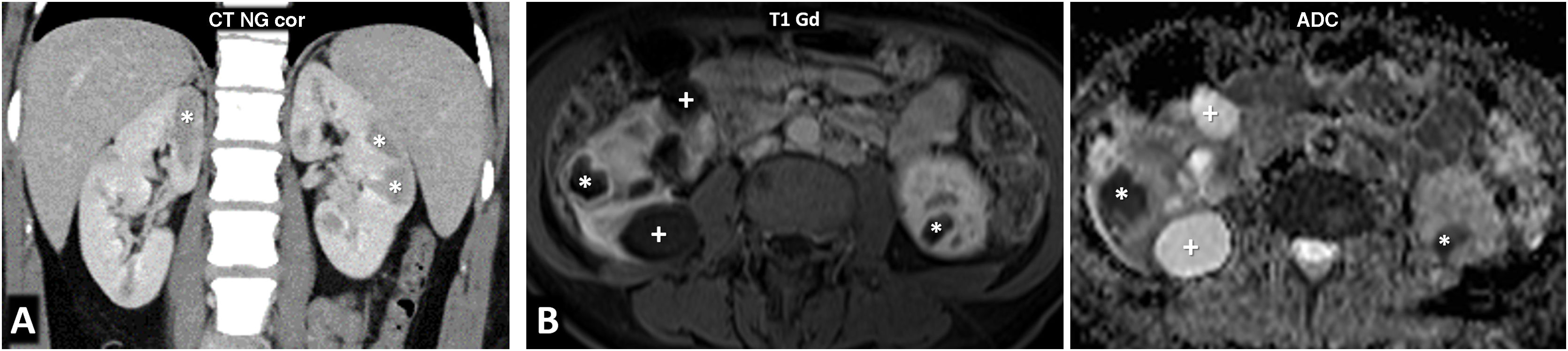
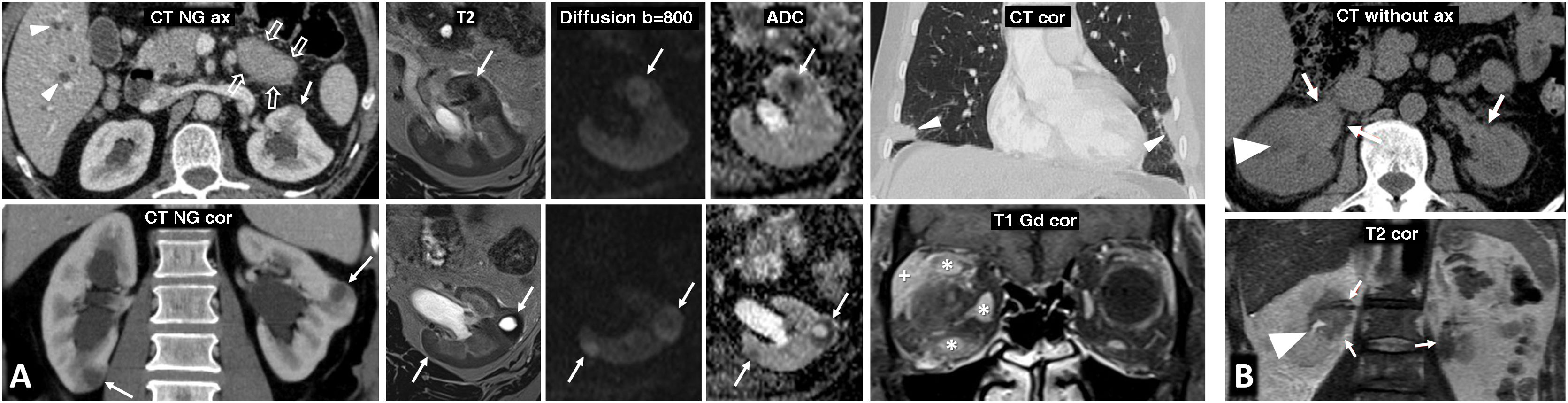
80% of renal carcinomas (RC) are diagnosed incidentally by imaging. 2–4% of "sporadic" multifocality and 5–8% of hereditary syndromes are accepted, probably with underestimation. Multifocality, young age, familiar history, syndromic data, and certain histologies lead to suspicion of hereditary syndrome. Each tumor must be studied individually, with a multidisciplinary evaluation of the patient. Nephron-sparing therapeutic strategies and a radioprotective diagnostic approach are recommended.
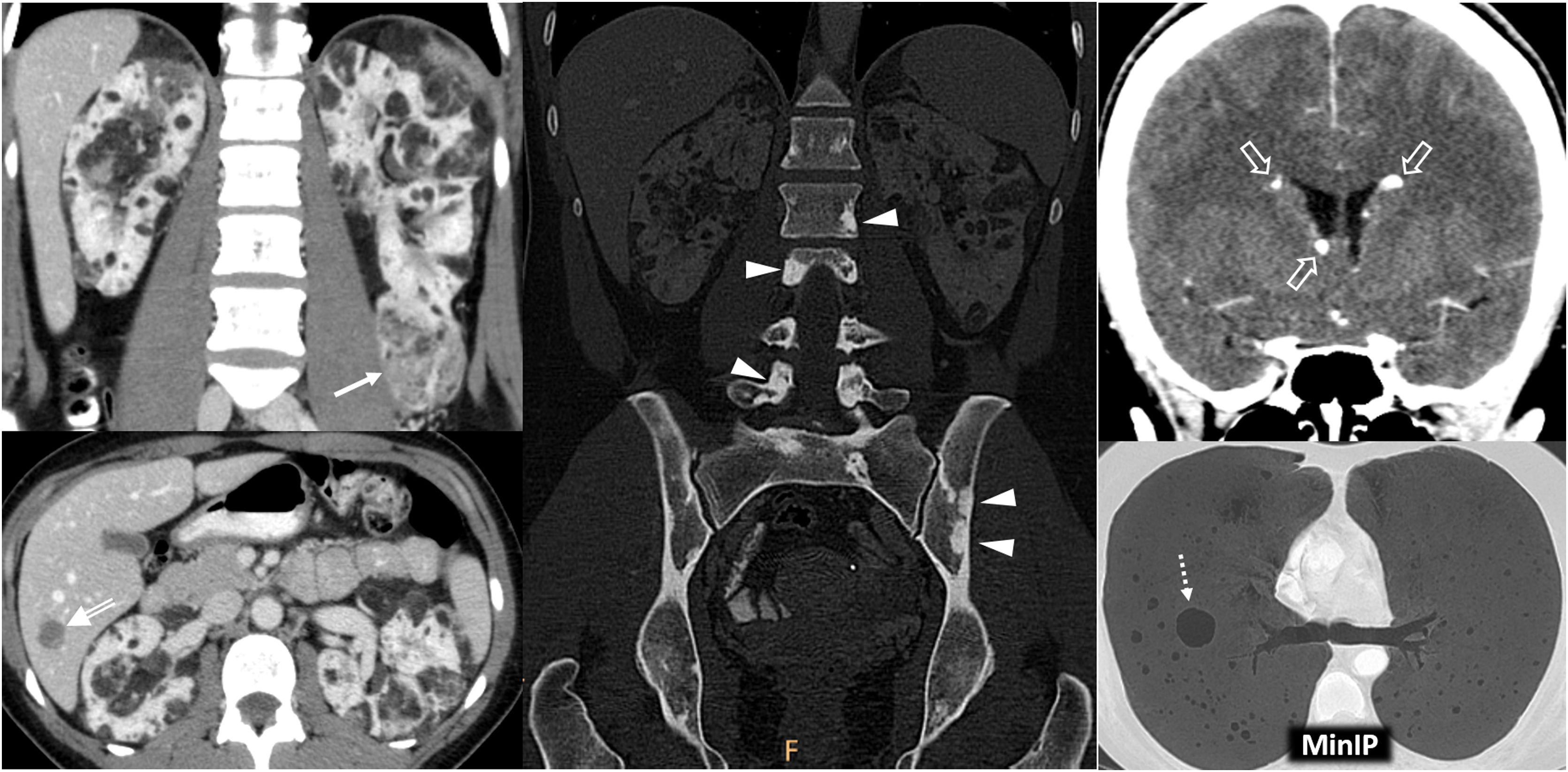
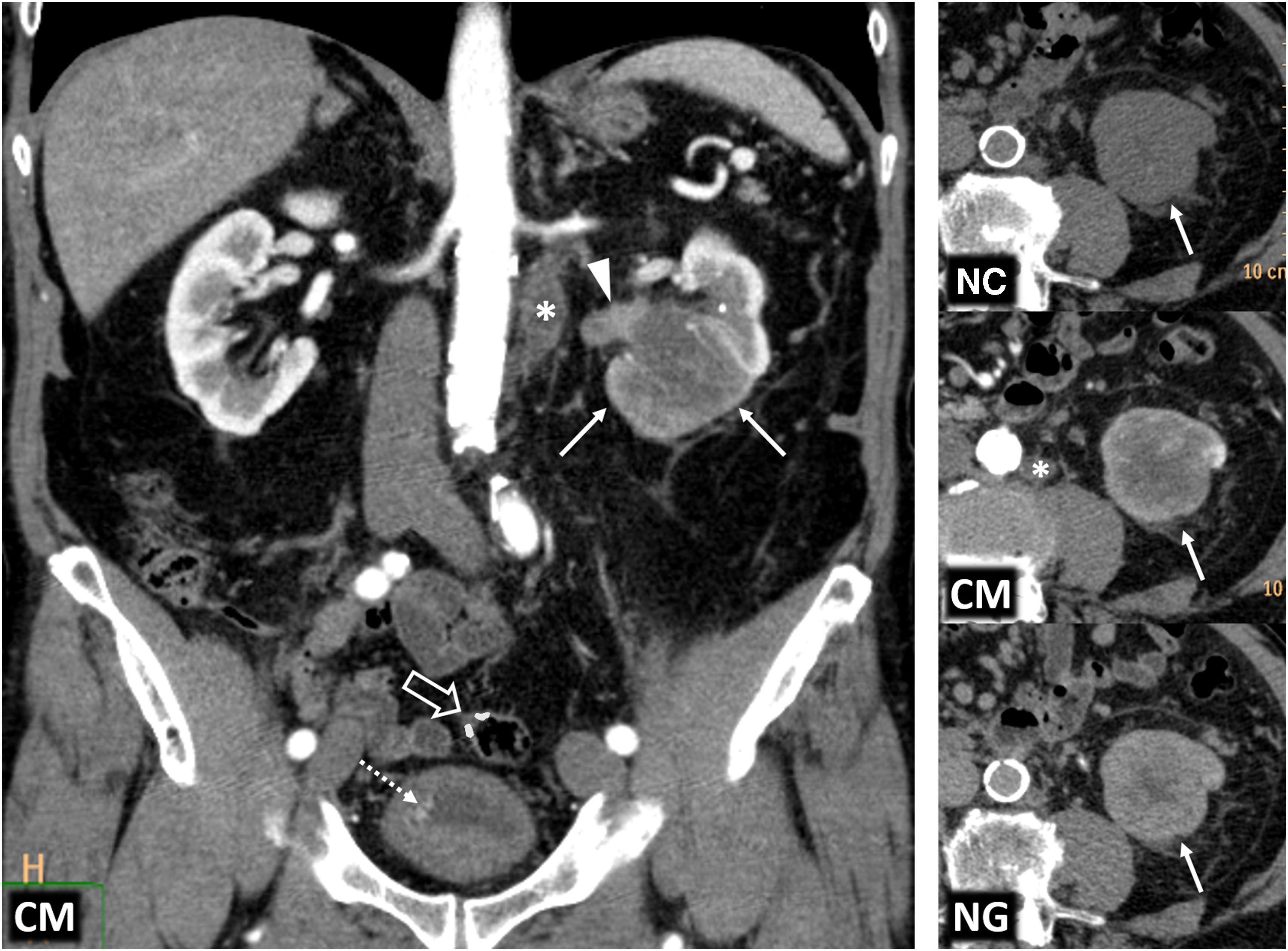
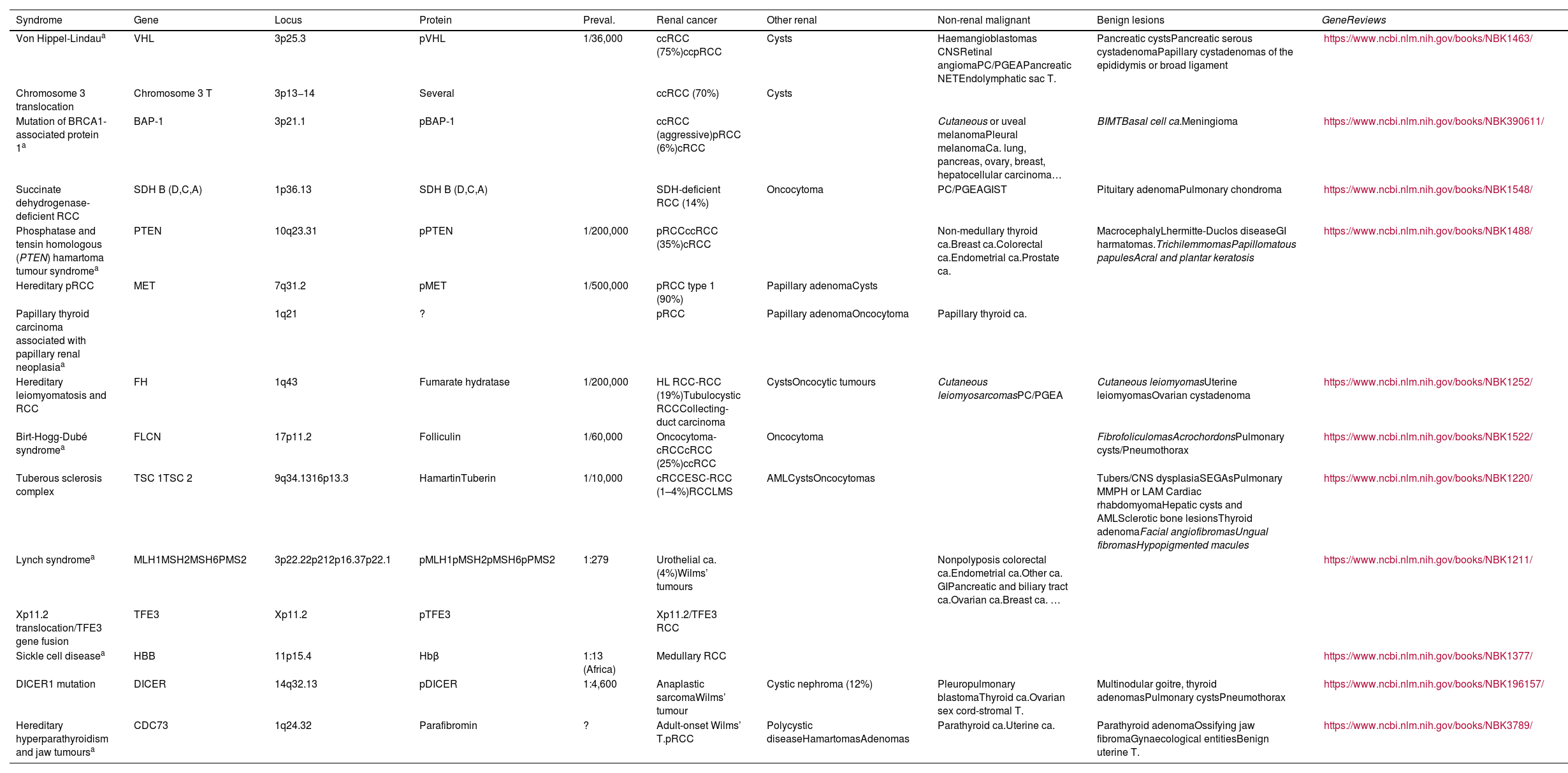
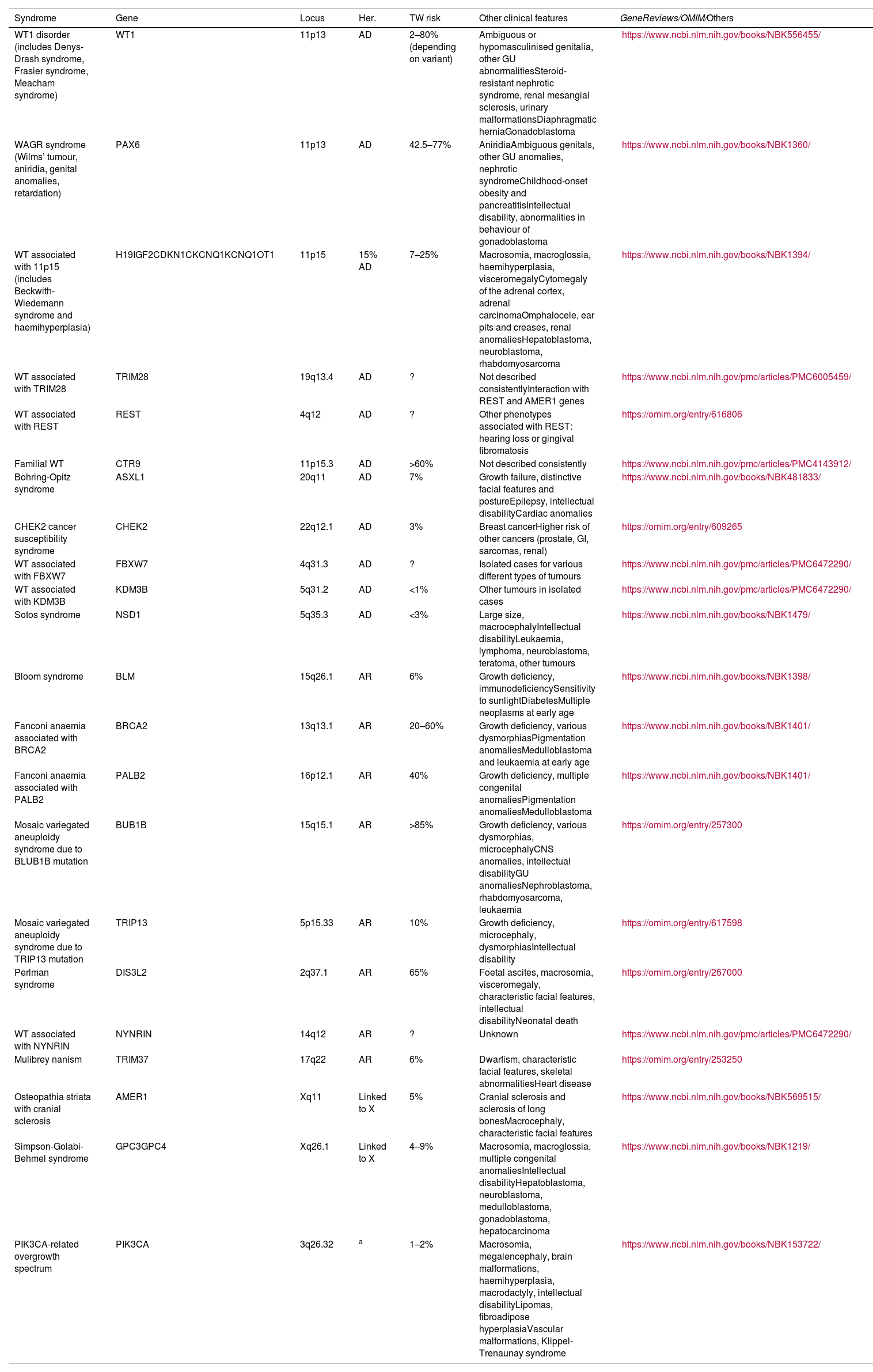
Relevant data for the radiologist in major RC hereditary syndromes are presented: von-Hippel-Lindau, Chromosome-3 translocation, BRCA-associated protein-1 mutation, RC associated with succinate dehydrogenase deficiency, PTEN, hereditary papillary RC, Papillary thyroid cancer- Papillary RC, Hereditary leiomyomatosis and RC, Birt-Hogg-Dubé, Tuberous sclerosis complex, Lynch, Xp11.2 translocation/TFE3 fusion, Sickle cell trait, DICER1 mutation, Hereditary hyperparathyroidism and jaw tumor, as well as the main syndromes of Wilms tumor predisposition.
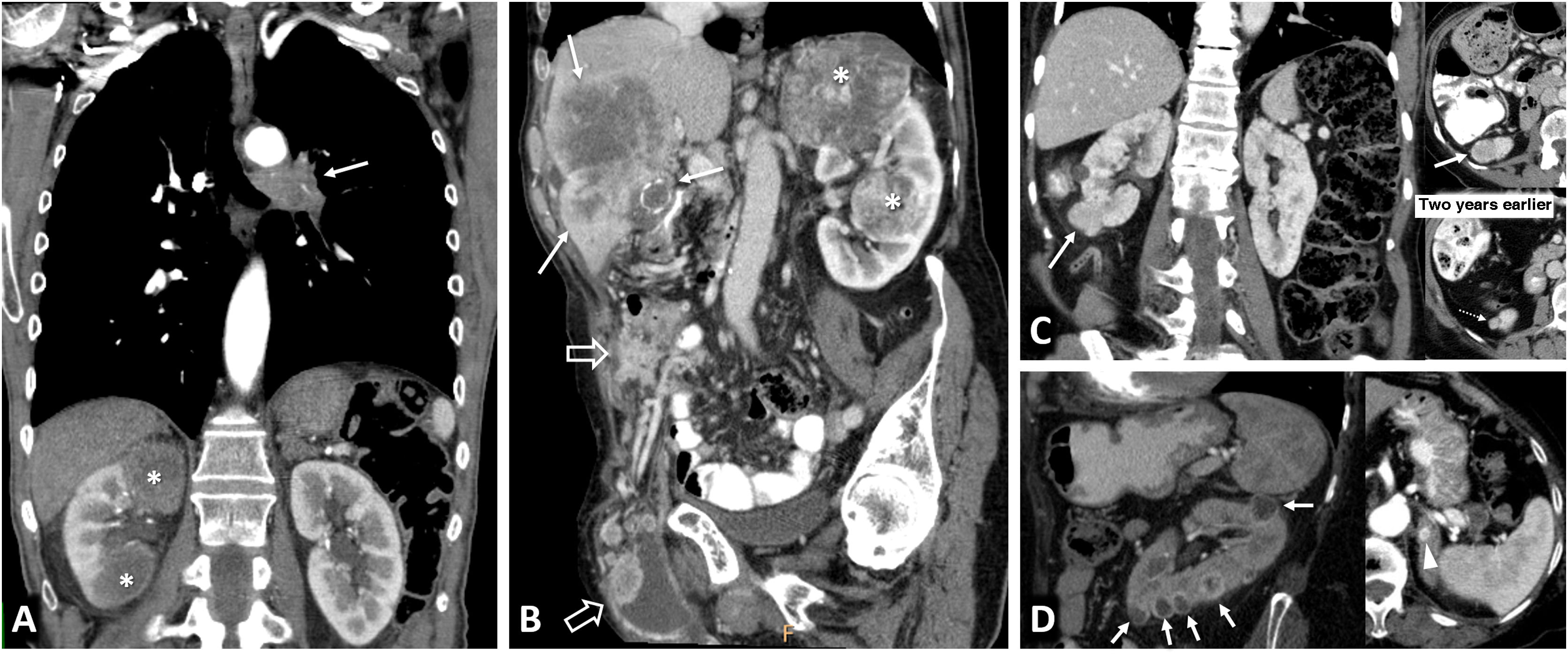
The concept of "non-hereditary" familial RC and other malignant and benign entities that can present as multiple renal lesions are discussed.
El 80% de los carcinomas renales (CR) se diagnostican incidentalmente por imagen. Se aceptan 2–4% de multifocalidad “esporádica” y 5–8% de síndromes hereditarios, probablemente con infraestimación. Multifocalidad, edad joven, historia familiar, datos sindrómicos y ciertas histologías hacen sospechar síndrome hereditario. Debe estudiarse individualmente cada tumor y multidisciplinarmente el paciente, con estrategias terapéuticas conservadoras de nefronas y abordaje diagnóstico radioprotector.
Se revisan los datos relevantes para el radiólogo en los síndromes de von Hippel-Lindau, Translocación de cromosoma-3, Mutación de proteína-1 asociada a BRCA, CR asociado a déficit en Succinato-deshidrogenasa, PTEN, CR papilar hereditario, Cáncer papilar tiroideo-CR papilar, Leiomiomatosis hereditaria y CR, Birt-Hogg-Dubé, Complejo esclerosis tuberosa, Lynch, Translocación Xp11.2/Fusión TFE3, Rasgo de células falciformes, Mutación DICER1, Hiperparatoridismo y tumor mandibular hereditario, así como los principales síndromes de predisposición a tumor de Wilms.
Se discuten el CR familiar “no hereditario” y otras entidades malignas y benignas que pueden presentarse como lesiones renales múltiples.

