


A família dos tumores de Ewing compreende um espectro de doenças malignas de células embrionárias neuroectodérmicas primitivas, que migram da crista neural. O sarcoma de Ewing primário do rim é uma neoplasia rara representando menos de 1% dos tumores renais e é caracterizado por um comportamento biológico altamente agressivo.
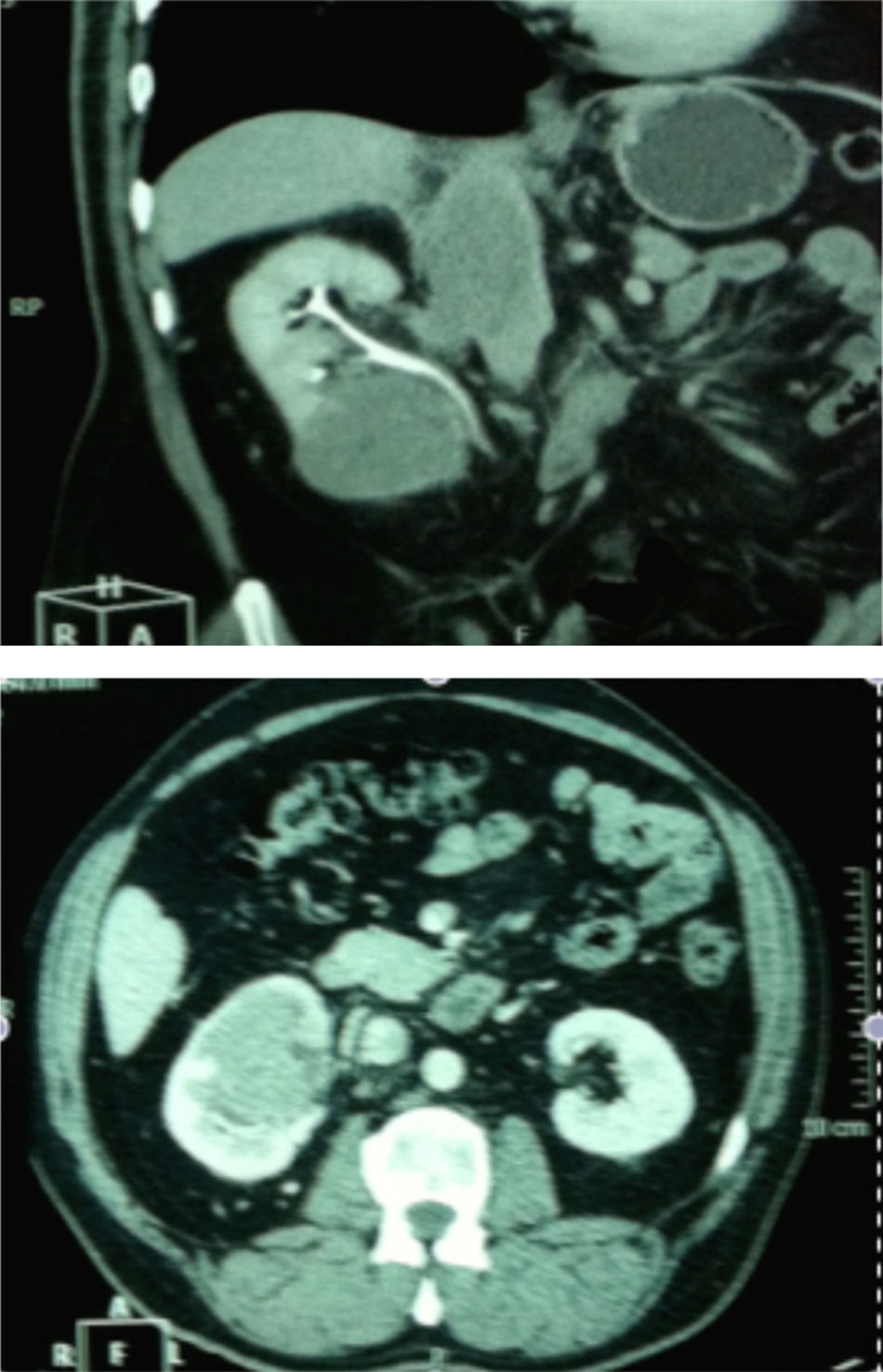
Apresentamos o caso de um homem de 53 anos, assintomático, a quem foi diagnosticada uma massa incidental do rim direito através de uma ecografia renal de rotina. O estudo foi complementado com TAC, que confirmou uma lesão sólida com 67x65mm, a ocupar a metade inferior do rim direito com invasão da veia renal, veia cava inferior e aurícula direita. O doente foi submetido a nefrectomia radical direita com exérese do trombo da veia cava inferior e aurícula direita sob circulação extracorpórea com paragem hipotérmica profunda. A histologia das peças operatórias revelou aspetos morfológicos e perfil imunoistoquímico sugestivo de sarcoma de Ewing. Cirurgia e pós‐operatório sem intercorrências. O paciente foi submetido a quimioterapia e faleceu 21 meses após a cirurgia por progressão da doença.
The Ewing's family of tumours comprises a spectrum of malignancies of primitive neuroectodermal cells: embryonic cells that migrate from the neural crest. 1Primary kidney Ewing's sarcoma is a rare neoplastic disease representing less than 1% of renal tumors and is characterized by highly aggressive biological behavior.
We reported a case of an asymptomatic, 53 year old man with an incidental lesion in the right kidney found in a routine renal ultrasound. Computed tomography showed a solid nodule with 67x65mm, occupying the lower pole of the right kidney with renal vein, inferior vena cava and right auricular invasion. The patient underwent a right radical nephrectomy and Inferior vena cava and atrial tumor thrombectomy with cardiopulmonary bypass and deep hypothermic circulatory arrest. Pathologic characteristics and immunohistochemical analysis confirmed the diagnosis of Ewing's Sarcoma. Surgery and early post‐operative were free of complications. The patient underwent chemotherapy and died 21 months after the surgery of relapse of the disease.
A família de tumores de Ewing compreende um espectro de neoplasias de células neuroectodérmicas primitivas, as quais são células embrionárias que migram da crista neural1. Os tumores pertencentes a esta família têm localização preferencial no osso, podendo atingir os tecidos moles das extremidades e mais raramente os órgãos viscerais2. O primeiro caso descrito deste tipo de tumores foi em 1918 por Arthur Stout e envolvia o nervo cubital3,4.
O sarcoma de Ewing (SE) e o tumor neuroectodérmico primitivo (PNET) foram inicialmente descritos como 2 entidades distintas. Atualmente, são considerados como tendo uma origem comum, baseada no facto de ambos terem a translocação cromossómica (11;22)(q24;12) em mais de 85% dos casos5.
Os SE de origem renal são uma entidade rara na população adulta e apresentam uma evolução agressiva2.
Apresentamos um caso de um SE primário do rim com trombo extenso na veia cava e aurícula direita, associado a uma breve revisão da literatura disponível.
Caso clínicoPaciente de 53 anos de idade, sexo masculino, não fumador, foi encaminhado para a consulta de urologia por uma lesão incidental do rim direito, diagnosticado em ecografia renal de rotina. Assintomático e sem antecedentes relevantes. A tomografia computorizada abdominal (fig. 1) mostrou uma lesão sólida com 67x65mm na metade inferior do rim direito com invasão do seio, veia renal, veia cava inferior (VCI) e aurícula direita, e cuja angiorressonância magnética confirmou o diagnóstico. O ecocardiograma transtorácico mostrava uma massa ecodensa, heterogénea, imóvel na aurícula direita, emergindo da VCI compatível com o trombo.
.")
O doente foi submetido a nefrectomia radical direita por via aberta, com exérese do trombo da VCI e aurícula sob circulação extracorpórea com paragem hipotérmica profunda, por uma equipa multidisciplinar – urologista, cirurgião vascular e cirurgião cardiotorácico.
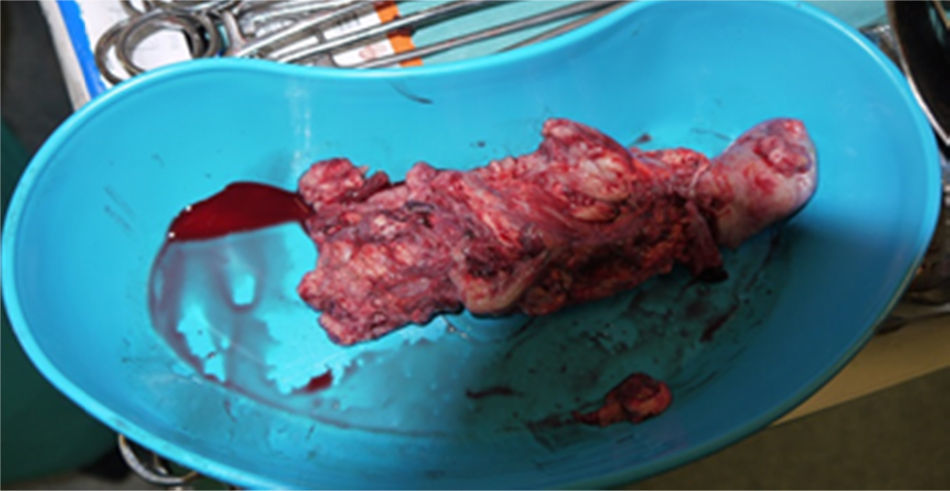
O pós‐operatório decorreu sem complicaçõesA histologia das peças operatórias (trombo [fig. 2] e tumor) revelou aspetos morfológicos e perfil imunoistoquímico sugestivo de SE.

A peça de nefrectomia radical direita revelou tumor de 9x8x7cm no polo inferior do rim invadindo a gordura adjacente à árvore pielocalicial, mas sem invadir a gordura perirrenal.
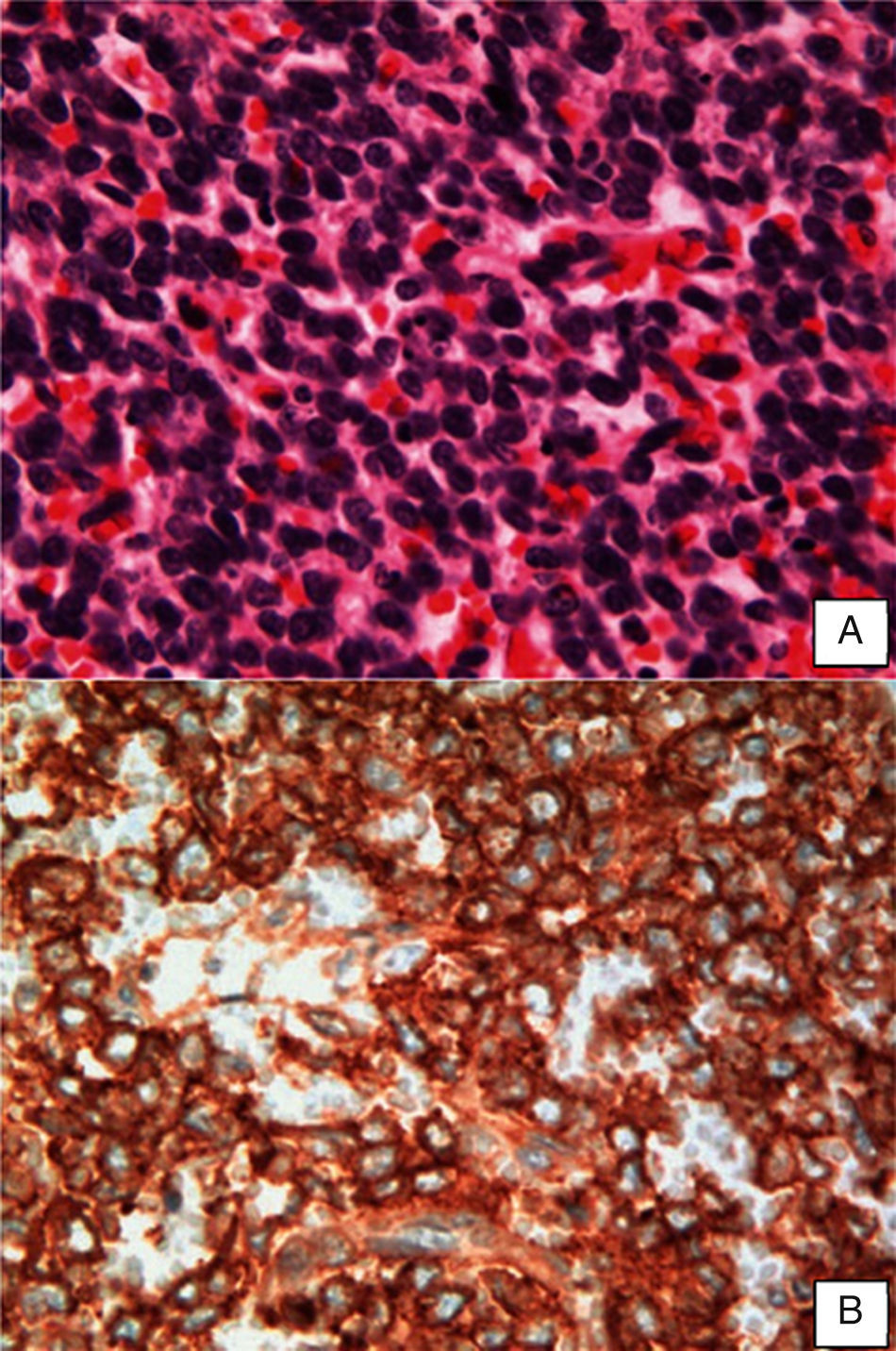
O exame histológico das peças cirúrgicas (fig. 3) mostrou uma população monótona de células de núcleo redondo e basofílico, com cromatina condensada e escasso citoplasma com numerosas áreas de necrose. O perfil de marcadores imunoistoquímica revelou células tumorais positivas para cluster of differentiation 99 (CD99) e enolasa neuronal específica (NSE), e negativas para vimentina, citoqueratina (CK), desmina, AML, CD34 e S100.

Exames de estadiamento mostraram um nódulo de 5,5mm no lobo inferior do pulmão esquerdo. A cintigrafia óssea foi negativa para depósitos ósseos secundários.
O paciente foi submetido a 4 ciclos de quimioterapia adjuvante com vincristina, adriamicina e ciclofosfamida, alternado com ifosfamida e ectoposido, com regressão do nódulo pulmonar em TC realizado 7 meses após a cirurgia.
Estável durante 10 meses, uma nova TC mostrou lesões secundárias no mediastino e pulmão compatíveis com progressão da doença. Iniciou quimioterapia de segunda linha com docetaxel e gemcitabina, mas acabou por falecer 4 meses depois.
DiscussãoO SE do rim é uma entidade rara e agressiva, com 200 casos publicados na literatura5. Ocorre em menos de 1% dos tumores renais. É um tumor mais frequente em crianças e jovens adultos, sendo o sexo masculino mais afetado com 3:16,7,4. A idade varia entre os 4‐69 anos4. A apresentação clínica é inespecífica e variável, desde ausência de sintomatologia, até casos de hematúria, massa palpável e dor no flanco ou sintomas derivados de metástases à distância.
O primeiro caso descrito de sarcoma de Ewing do rim foi em 1975, por Seemayer et al.7,8A suspeita diagnóstica inicial é de tratar‐se de um tumor do rim, sendo o mais frequente carcinoma de células claras, não havendo diferenças entre eles em exames de imagem. O diagnóstico definitivo é habitualmente feito após a cirurgia, recorrendo a técnicas de imunoistoquímica e moleculares. Macroscopicamente, os PNET são tumores grandes, volumosos e tendem a ser de cor acinzentada, encapsulados, com áreas focais de hemorragia e necrose. Histologicamente, apresentam pequenas células redondas com núcleo e escasso citoplasma. Podem exibir diferentes padrões de «rosetas» e elementos fusiformes5,9–11. O diagnóstico diferencial de tumores do rim com morfologia de pequenas células redondas faz‐se com outras neoplasias não relacionadas, e cujo tratamento e prognósticos são distintos7,12. Estão incluídas o linfoma maligno, rabdomiosarcoma embrionário, neuroblastoma renal, tumor de Wilms, osteosarcoma de pequenas células, tumor desmoplástico de pequenas células, sarcoma sinovial e tumor neuroendócrino de pequenas células10,11.
Os marcadores CD99, NSE e anticorpos monoclonais podem ajudar para o correto diagnóstico. CD99 é positivo nos tumores da família de SE, mas não pode ser utilizado como marcador absoluto, por não ser específico e positivar nos carcinomas de células pequenas e linfomas. NSE, neurofilamentos S100 e cromogranina‐A podem excluir SE extraósseo6,11,13. O diagnóstico definitivo pode ser confirmado através de análise citogenética pela existência da translocação t(11;22)(q24:q12), que parece ser única a este tipo de tumores, sendo positiva em 85% dos casos12–14.
O SE renal tem um comportamento agressivo, com cerca de 30% dos diagnósticos iniciais apresentarem metástases. Os locais mais frequentes de localização secundária são os gânglios, pulmão, fígado e osso13,15.
A taxa de sobrevivência livre de doença a 5 anos após cirurgia é de 45‐55% e o prognóstico melhora nos doentes com doente localizada11,15. Estudos sugerem que o SE extraósseo pode ser um fator preditor de mau prognóstico, assim como extensão intravascular, idade avançada, metástases no diagnóstico inicial, ressecção incompleta da lesão e má resposta a quimioterapia8,13.
O tratamento desta doença baseia‐se na cirurgia radical associada a quimioterapia e radioterapia. O papel da radioterapia não é claro, mas pode ser utilizado na doença localmente avançada e envolvimento da fáscia de Gerota8,14,16.
A quimioterapia adjuvante melhora o prognóstico deste tipo de tumores. Estudos retrospetivos mostram uma boa resposta a quimioterapia, com uma taxa de resposta global de 66% e um benefício clínico de 75%12. No entanto, estes estudos apresentam várias limitações, desde a natureza retrospetiva dos mesmos e do reduzido número de doentes utilizados para calcular as diferentes variáveis, que podem desta forma aumentar o risco de viés. Os pacientes receberam diferentes esquemas de quimioterapia e não houve critérios uniformes na interrupção dos mesmos. Não existe um esquema pré‐definido. Os fármacos mais frequentemente utilizados são a adriamicina, etoposido, dactinomicina, vincristina, ciclofosfamida e ifosfamida6,7,17,18. As guidelines existentes, nomeadamente da National Comprehensive Cancer Network (NCCN)19, referem‐se a tumores da família de SE de qualquer localização e dividem as diferentes opções de tratamento quimioterápico entre doença localizada e doença metastizada.
Neste caso, o doente realizou 2 linhas de quimioterapia indicadas nas guidelines da NCCN para doença metastizada19.
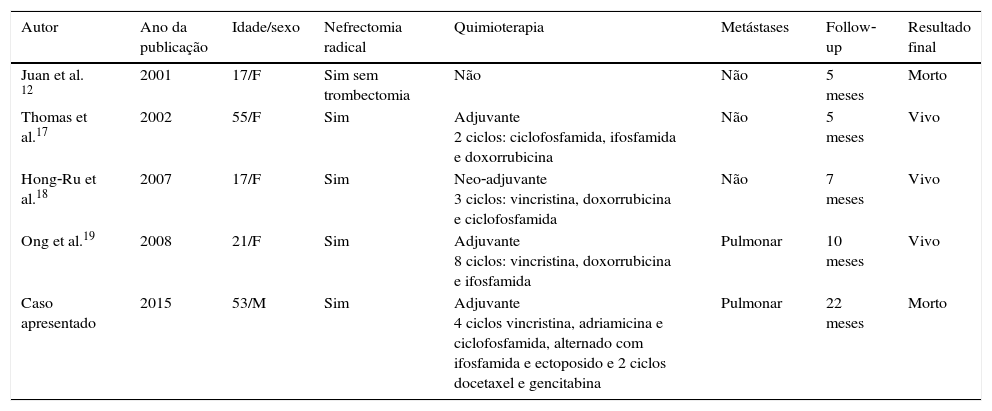
Na literatura, existem apenas 5 casos de SE primário do rim com trombo estendendo‐se até à aurícula direita (tabela 1).
Revisão na literatura de sarcoma de Ewing do rim com trombo na aurícula
| Autor | Ano da publicação | Idade/sexo | Nefrectomia radical | Quimioterapia | Metástases | Follow‐up | Resultado final |
|---|---|---|---|---|---|---|---|
| Juan et al. 12 | 2001 | 17/F | Sim sem trombectomia | Não | Não | 5 meses | Morto |
| Thomas et al.17 | 2002 | 55/F | Sim | Adjuvante 2 ciclos: ciclofosfamida, ifosfamida e doxorrubicina | Não | 5 meses | Vivo |
| Hong‐Ru et al.18 | 2007 | 17/F | Sim | Neo‐adjuvante 3 ciclos: vincristina, doxorrubicina e ciclofosfamida | Não | 7 meses | Vivo |
| Ong et al.19 | 2008 | 21/F | Sim | Adjuvante 8 ciclos: vincristina, doxorrubicina e ifosfamida | Pulmonar | 10 meses | Vivo |
| Caso apresentado | 2015 | 53/M | Sim | Adjuvante 4 ciclos vincristina, adriamicina e ciclofosfamida, alternado com ifosfamida e ectoposido e 2 ciclos docetaxel e gencitabina | Pulmonar | 22 meses | Morto |
No único caso em que não foi possível realizar a trombectomia, o doente acabou por falecer prematuramente. Os outros 4 casos foram submetidos a cirurgia radical com exérese do trombo associados a quimioterapia adjuvante. Como já foi referido anteriormente, existem várias opções de esquemas de quimioterapia possíveis, dependendo da existência de doença metastática no diagnóstico inicial. Estes casos mostram a grande variabilidade no tratamento, quer seja no número de ciclos ou no esquema terapêutico. Os artigos encontrados têm pouco tempo de seguimento (média de 6,75 meses), o que pode alterar o que se pensa da história natural da doença. No entanto, há uma tendência nos vários artigos revistos em mostrar a agressividade destes tumores e a necessidade de tratamento multimodal.
Em conclusão, trata‐se de um tumor raro, de comportamento agressivo e mau prognóstico, e que deve constar sempre no diagnóstico diferencial de massas renais, sobretudo em jovens adultos. O tratamento deve ser agressivo com cirurgia associada a quimioterapia adjuvante, que podem melhorar a sobrevivência a longo prazo.
Responsabilidades éticasProteção dos seres humanos e animaisOs autores declaram que os procedimentos seguidos estavam de acordo com os regulamentos estabelecidos pelos responsáveis da Comissão de Investigação Clínica e Ética e de acordo com os da Associação Médica Mundial e da Declaração de Helsinki.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram que não aparecem dados de pacientes neste artigo.
Conflito de interessesOs autores declaram não haver conflito de interesses.
