






Chronic granulomatous disease (CGD) is a disorder of phagocyte function, characterized by pyogenic infections and granuloma formation caused by defects in NADPH oxidase complex activity. Although the effect of CGD mainly reflects the phagocytic compartment, B cell responses are also impaired in patients with CGD.
Materials and methodsFlow cytometric analysis was performed on peripheral blood samples from 35 CGD patients age-matched with healthy controls (HC). The target cells of our study were the naive (IgD+/CD27−), memory (IgD−/CD27+), and B1a (CD5+) cells. Immunoglobulins (Igs) were also measured. This study was performed in a Latin American cohort.
ResultsWe found significantly higher levels of naive B cells and B1a cells, but lower levels of memory B cells were found in CGD patients compared to HC. There was no significant difference of cell percentages per inheritance type.
DiscussionOur findings suggest that the deficiency of NADPH oxidase components can affect the differentiation of naive B cells to memory B cells. Consequently, memory cells will be low, which also influenced the expression of CD27 in memory B cells and as a result, the percentage of naive cells increases. An altered phenotype of B lymphocytes in CGD patients may contribute to the opportunistic infections and autoimmune disorders that are seen in this disease.
Chronic granulomatous disease (CGD) is a primary immunodeficiency disorder that is caused by an inherited defect in any one of five subunits of phagocyte-derived nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. Through it, the phagocytic cells generate microbicidal superoxide and its metabolites hydrogen peroxide, hydroxyl anion, and hypohalous acid.1
The CYBB gene encoding the enzymatic center of the NADPH oxidase complex, gp91phox, is on the X chromosome, and mutations in this gene account for approximately 60–80% of cases. Autosomal forms of CGD result from mutations in genes from p47phox, p67phox, p22phox, or p40phox, whereas p47phox.1
In this condition, there is a profound defect in the respiratory burst that normally accompanies phagocytosis, consequently conferring an increased risk of bacterial and fungal infections.2,3 Elevated autoantibodies and hyperinflammatory and autoimmune manifestations, such as systemic lupus erythematous, discoid lupus, pneumonitis, and inflammatory bowel disease are associated with CDG. Treatment is based on antimicrobial prophylaxis, and hematopoietic stem cell transplantation (HSCT) is one of the curative therapies.4
As part of immune reconstitution studies in the context of non-myeloablative HSCT for CGD, several CGD patients were observed to display a substantial decrease in peripheral blood B cells expressing CD27 before undergoing HSCT.5
Bleesing et al. studied the B cell compartment in patients with CGD. They found a profound reduction of B cells expressed in the memory B cell marker, CD27, and an expansion of CD5+ cells (B1a cells). Mohsenzadegan et al. reproduced the same results in 31 patients with CGD.6,7
B cells have also been shown to produce superoxide in response to various stimuli, but these B cells produce only a small amount of superoxide even when fully stimulated.7 B lymphocytes use their slowly generated O2− and H2O2 for other purposes, such as acting as second messengers in signal transduction pathways.8
The finding of reduced CD27+ B cells in CGD patients may be linked to the observation that B cells possess a superoxide generating system with structural homology to the NADPH oxidase system of phagocytes, but with 50–100-fold less oxidase activity. A role for the NADPH oxidase system in B cells is suggested by oxidase activity in response to surface Ig cross-linking and the prevention of proliferation of human peripheral blood B cells by interference with oxidase activity.6
It is not clear whether chronic inflammatory manifestations and autoimmune diseases in CGD are secondary to altered superoxide production, or whether a defect in adaptive immune system, such as a defect in B lymphocytes, is involved.9
We aimed to study the phenotype description of the B cells in peripheral blood of patients with CGD in a Mexican population using CD27+ B cells (as memory B cell markers), IgD+/CD27− (as naive B cell markers), and B1a cells defined by CD5 expression, which always contrasts with the healthy group. The results revealed a profound reduction in CD27+ memory cells, and an increased count of naive cells. These seem to suggest that the deficiency of NADPH oxidase components can affect the differentiation of naive B cells to memory B cells. It is the first time this study has been performed in a Latin population.
Materials and methodsSubjectsThirty-five patients with CGD (CGD group) were age-matched with healthy controls (healthy control (HC) group); both gender donors participated, with the mean age of 7.2 years old (age range of 0.5–37 years). All patients were previously diagnosed as CGD with flow cytometry using dihydrorhodamine 123 (DHR) assay. We reviewed the file of each patient and registered if they had had, at some time of the disease, an inflammatory entity as granuloma, autoimmunity or hemophagocytic lymphohistiocytosis, we also recorded if the autoimmunity event had resolved or not. The patients were recruited from the Immunodeficiencies Research Unit at the National Institute of Pediatrics in Mexico City from 2016 to 2018. The Research and the Ethical Committees approved the study. All these patients were registered in the Latin American Society for Immunodeficiencies platform (registrolasid.org). Informed consent and/or assent was obtained from all subjects or their parents.
Flow cytometryAll patients were previously diagnosed as CGD with flow cytometry using DHR 123 assay as described by Vowells et al. in 1995.9
Lithium heparin tubes were used. Three milliliters of peripheral blood specimens were obtained from patients and healthy controls by phlebotomy on site or were mailed in and studied the next day.
B cell subpopulation assessmentBlood specimens were stained with the mAbs: PE-labeled anti-human CD19, PerCP Cy5 labeled anti-human CD5, APC labeled anti-human CD27, and PE-Cy7 labeled anti-human IgD. All mAbs were obtained from BD Pharmigen and used as recommended by the manufacturers. We defined memory cells as CD19+/IgD−/CD27+, naive cells CD19+/IgD+/CD27−, and B1a cells (CD19+/CD5+). We acquired 10,000 labeled cells by flow cytometry simultaneously in the patients and HC samples. We used FACS ARIA I and FACS Diva Software version 4.0.1. For the analysis, we used FlowJo software version 8.7. In it, lymphocytes were selected in a gate by drawing a region around the cell population on the forward scatter/side scatter dot plot. Surface marker expression was assessed through the relative number of positive cells. Isotype control antibodies were used to separate positive and negative cells on PE, PerCP-Cy5, and PerCP-Cy7 and APC fluorescence channels.
Immunoglobulin measurementImmunoglobulin serum levels of CGD patients were determined by the nephelometric method. The patients’ immunoglobulin levels (IgG, IgM, and IgA mg/dl) in the patients were compared with the normal ranges according to age groups.
Statistical analysisAll data were analyzed using SPSS statistical software package version 25 (SPSS Inc., Chicago IL, USA). Comparisons between medians were performed by Wilcoxon matched-pairs signed rank test or Mann–Whitney test where appropriate. A level of p<0.05 was considered significant for all tests. All the results had a 95% confidence interval (CI).
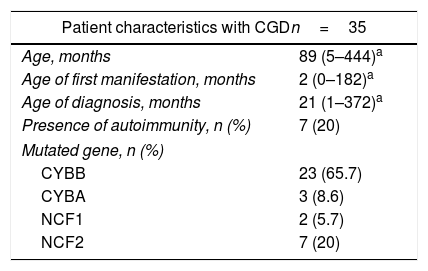
ResultsPatient characteristicsThis study was conducted on 35 CGD patients age-matched with HCs; by gender, 27 (77.1%) male and eight (22.9%) female patients were included. When the sample was extracted, only one patient had an active infection, the rest of the patients were clinically without infection. The median age of the group was 7.4 years (range, 0.5–37 years). By type of inheritance, 23 patients (65.7%) were found with the X-linked form (CYBB) and 12 patients (34.3%) with CGD AR; within the AR group, seven patients (20%) had an NCF2 mutation, three (8.6%) had CYBA and two (5.7%) had NCF1. Eight percent of the total of patients had the novo mutations. One (2.8%) female patient had X chromosome inactivation (Table 1).
General characteristics of the 35 studied patients.
CGD patients displayed a significantly decreased lymphocyte count with 24.7% (95% CI: 23.2–30.9), versus 34% (95% CI: 28.5–36.3) in S, with p=0.030 (Fig. 1A). The percentage of B cells was statistically significantly increased in the HC group by 16.6% (95% CI: 16–19) compared to the CGD group, which had 14.4% (95% CI: 12.4–17.8), with p=0.038 (Fig. 1B). Regarding B1a cells (CD19+CD5+), a significantly high percentage of 71.5% (95% CI: 66.5–76) with p=0.012 was found in CGD patients, in contrast with the HC results of 58.7% (95% CI: 47.6–65.7) (Fig. 2A). The median fluorescence intensity of CD5+ cells in the CGD group were 100 (95% CI: 90.9–120) versus 117.5 (95% CI: 106–150); however, this was not statistically significant (p=0.350, Fig. 2B).
 percentage of lymphocyte population of CGD group and HC; also, (B) percentages of B cells are shown. p=values were determined with the non-parametric Mann–Whitney test. CD19+ cells established the B-cell population. Chronic granulomatous disease (CGC), healthy controls (HC).")
The lymphocyte and B cell phenotype found in CGC group and healthy controls; (A) percentage of lymphocyte population of CGD group and HC; also, (B) percentages of B cells are shown. p=values were determined with the non-parametric Mann–Whitney test. CD19+ cells established the B-cell population. Chronic granulomatous disease (CGC), healthy controls (HC).
. The MFI of CD5+ cells in the CGD and HC groups (B). p=values were determined with the non-parametric Mann–Whitney test. Chronic granulomatous disease (CGC), healthy controls (HC), median fluorescence intensity (MFI).")
Percentage of expansion of B1a CD19+CD5+ cells in the CGD and HC groups (A). The MFI of CD5+ cells in the CGD and HC groups (B). p=values were determined with the non-parametric Mann–Whitney test. Chronic granulomatous disease (CGC), healthy controls (HC), median fluorescence intensity (MFI).
The CGD group showed a highly significant reduction of 3.8% (95% CI: 2.6–12.2) in the percentage number of peripheral blood B cells that expressed memory (IgD+CD27−) versus 14.6% (95% CI: 13–21.4) of the HC group with p≤0.001 (Fig. 3A). On the other hand, a significantly increased number of naive cells (IgD+CD27−) were exhibited, 85.9% in CGD patients (95% CI: 75.1–87.2) versus 77.9% (95% CI: 65–77.3) of HC with p=0.003 (Fig. 3B). Moreover, the switched memory cells (IgD− CD27+) of CDG had a decreased percentage of 4.5% (95% CI: 4–6.5) regarding HC with 5% (95% CI: 5.6–10.2) with p=0.015. Double negative cells (IgD−CD27−) were found to be higher in the CGD group at 3.7% (95% CI: 2.9–9.3) versus 1.93% (95% CI: 1.6–4.8) in the HC group with p=0.005.
 Percentage of memory cells expressed (IgD+CD27+) in CGD and HC groups, and (B) percentage of naive cells expressing (IgD+CD27−) in CGD and HC groups are shown. p=values were determined with the non-parametric Mann–Whitney test. Chronic granulomatous disease (CGC), healthy controls (HC).")
The B-cell phenotype of patients with CGD is characterized by lower memory and higher naive cell counts. (A) Percentage of memory cells expressed (IgD+CD27+) in CGD and HC groups, and (B) percentage of naive cells expressing (IgD+CD27−) in CGD and HC groups are shown. p=values were determined with the non-parametric Mann–Whitney test. Chronic granulomatous disease (CGC), healthy controls (HC).
The inherence the X-linked and autosomal recessive forms of the B cell subsets of the CGC group were also compared; no statistically results were found. The lymphocyte percentage of the X-linked group was 25.9 (95% CI: 13.4–59.6) and 21.2% (95% CI: 17.7–29.8) in AR forms, with p=0.161. The B cell percentage of the X-linked group was 14.4 (95% CI: 4.6–32.7) and 14.3% (95% CI: 10.5–20.7) in AR forms with p=0.873. In the X-linked group, naive cells IgD+CD27− were 83.9% (95% CI: 70.7–96.4) and 89.6% (95% CI: 7.7–95.5) in AR with p=0.164. XL memory cells (IgD+CD27+) were 4.6 (95% CI: 0.69–21) and 2.9% (95% CI: 0.54–81) for AR, with p=0.195 (Fig. 4A). X-linked switched memory cells (IgD−CD27+) were 5.4 (95% CI: 0.03–14.8) and 3.8% (95% CI: 0.55–12.6) for AR, with p=0.085. Double negative cells in X-linked patients were 3.7 (95% CI: 0–10.5) and 3% (95% CI: 1.15–52.5) with p=0.644. Expansion B1a CD19+CD5+ cells in X-linked patients were 75.2 (95% CI: 38.1–93.4) and 66.3% (95% CI: 50.7–86.2) for AR, with p=0.062 (Fig. 4B). The median fluorescence intensity of CD5+ was 119.5 (95% CI: 31–193) in X-linked patients and 79.2 (95% CI: 27.1–170) in the AR group, with p=0.068.
Autoimmunity in CDG patients and the B1a CD19+CD5+ cells percentage (B) in the CGD group by type of inheritance. p=values were determined with the non-parametric Mann–Whitney test. Autosomic recessive form (AR), X-linked form (XL).")
The relationship between CGD and hyperinflammatory and autoimmune disorders is well known. In this study, six patients (17%) were diagnosed with a concomitant autoimmune disease. Two cases (5.7%) with hemolytic anemia and two cases with inflammatory bowel disease were encountered. One case of each of the following entities (2.8%) was found: Kawasaki disease, discoid lupus erythematous, autoimmune thrombocytopenia, and autoimmune uveitis. All the patients had a favorable response to each one of the autoimmune diseases. The comparison of the percentage of B1a cells (CD19+CD5+) in patients with and without autoimmunity was 72.2 (59.2–92.1 median) and 71.8% (38.1–93.4 median), with p=0.643, respectively. An increased percentage of these cells in the positive autoimmunity group was expected based on the literature; nevertheless, the data were found to lack statistical significance.
Immunoglobulin serum levelsThe immunoglobulins of patients with CGD was previously reported to be high.10 This was also corroborated by our study in which, coinciding with the literature reports, high levels of serum immunoglobulins were found in all CGD patients (considering the age reference values); all the healthy subjects had normal immunoglobulins values11; for the patients the median IgG was 1527mg/dl (940–2700), IgM was 186.5mg/dl (71–341), and IgA was 208mg/dl (60–693).
DiscussionOur study showed that the B cell subset was altered in the CGD group, indicating a significant increase in B1a (CD19+CD5+) cells, naive cells (IgD− CD27+), and immunoglobulins (IgG, IgM, IgA). On the other hand, we found a significant decrease in peripheral lymphocytes and memory cells (IgD+ CD27+).
Regarding the expansion of B1a (CD19+CD5+) cells, our findings coincided with previous studies of the B cell compartment in CGD patients, in which high levels of this type of cells were also encountered.1,9
B1a cells (CD19+CD5+) are considered to be elements of the innate immune system. It is the main source of interleukin (IL)-10 that plays an important protective anti-inflammatory role and is required to prevent bacterial dissemination and host morbidity by controlling effector T cells. They are also an important source of natural IgM, which, in addition to its antimicrobial properties, helps maintain tissue homeostasis by cross-reaction with epitopes expressed on dead and dying cells.12–14
They are also an important component of barrier immunity, as they preferentially class switch to IgA to control microbes at mucosal surfaces. This immune regulation therefore functions as protection of the host against potentially fatal excessive pro-inflammatory responses, whereas it can also benefit the bacterium by dampening local pro-inflammatory responses, thus facilitating bacterial persistence.15
Shimomura et al. demonstrated that B1 cells could play a regulatory role in the development of colitis in a murine model by means of the increase in the generation of natural IgM production in response to microbial flora. There is also evidence of the production of immunomodulatory molecules by B1 cells, such as IL-10, GM-CSF, IL-3, and IL-35, which regulate inflammatory diseases. Therefore, patients with inflammatory bowel disease presented a low percentage of B1a cells, in contrast with our findings, in which CGD patients with inflammatory bowel disease had an increased percentage of CD5+ cells. Interestingly, the CD5+B1 subset tended to be elevated in patients with a polysystem autoimmune disease; there are examples in both human and mouse models of an association between autoimmune disease and B1 cells.14 In humans, elevated numbers of B1 cells have been reported in patients with CGD, in which it is already known that an increased activity of caspase-1 and increased release of IL-1b and other pro-inflammatory cytokines by activated mononuclear cells contributes to dysregulated inflammatory responses, even in the absence of clinical infection.15 This may promote the high incidence of autoimmune diseases. In mice, increased numbers of B-1 cells have been observed in naturally occurring and genetically manipulated strains that develop autoimmune manifestations, such as lupus erythematous disease and autoimmune hemolytic anemia. It is therefore possible that the induction of the B-1 phenotype serves to tolerate B cells expressing certain specificities while still keeping them available for certain responses.16Alternatively, susceptibility to autoimmune disease might result from diminished negative regulation of B-1 cells. As a result, B-1 cells producing low-affinity autoantibodies would receive T cell help, enter germinal centers, class switch, undergo somatic mutation, and as a result of affinity maturation, produce high affinity IgG autoantibodies.17
Concerning memory cells (IgD+CD27+), we found a significant decrease, and the number of naive cells (IgD+CD27−) was found to be high, always compared to HC; these results coincide with those of previous studies.6,18
In normal circumstances, after antigen-independent development in the bone marrow, immature B cells leave the bone marrow and gather in the longer-lived mature, naive (IgD+CD27+) B-cell pool. When these cells are stimulated by antigens in the presence of the appropriate costimulation, they will engage in a germinal center (GC) reaction and develop into plasma cells or memory B cells.19 Mohsenzadegan et al. suggested that deficiency of NADPH oxidase components can affect the differentiation of naive B cells to memory B cells and especially influence the expression of CD27 on memory B cells, and consequently the percentage of naive cells increases. It is also known that the NADPH oxidase complex, especially the p47phox protein, may be involved in other functions besides the production of superoxide, such as in the signaling and activation of some transcription factors in B cells.7 The development of memory B cells is essentially linked to the formation of GC in secondary lymphoid organs, so the significantly reduced number of these cells strongly support that GC reactions are disturbed in CGD. The reduced CD40L expression in T lymphocytes as well as CD40 expression on neutrophils has been shown in CGD patients. There is a connection between O2− generated by NADPH oxidase and CD40L induction. Deficiency in productions of NADPH oxidase in CGD patients might therefore indirectly affect the production of memory B cells, impair differentiation of naive B cells to memory B cells, and cause reduced memory B cells.9
In our cohort of patients with CGD, all patients had hypergammaglobulinemia. This condition has been related to autoimmune/autoinflammatory disorders, but its etiology remains unknown.
In summary, our study showed an altered B cell subset, significant increase in B1a cells, decrease in peripheral lymphocytes and memory cells, and hypergammaglobulinemia in the CGD group. This may be the result of the deficiency of NADPH oxidase complex that consequently disturbed the development of B cells in germinal centers and affects the differentiation of naive B cells to memory B cells. The increase of the B1a cells may be related to the pro-inflammatory and autoimmune state in CGD.
FundingThis work was supported by a grant from the Latin American Society of Immunodeficiencies (LASID). MA Yamazaki-Nakashimada, SE Espinosa-Padilla, BE del Rio-Navarro and L Blancas-Galicia have SNI-CONACYT fellowships.
Conflict of interestThe authors have no financial conflict of interest.
The authors thank the patients and healthy volunteers for their participation and the different clinic teams for their help in patient care and blood sample collection.
