


Chronic granulomatous disease (CGD) is an inherited disease that results from a defect in the phagocytic cells of the immune system. It is caused by defects in one of the major subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. The clinical presentations of CGD patients are heterogeneous.
ObjectivesThis is the first report from Egypt discussing clinical and laboratory data of twenty-nine patients (from 26 families) with CGD from a single tertiary referral centre.
ResultsThere were twenty male and nine female patients. The consanguinity rate was 76% (19/25). Their age of diagnosis ranged from 2 to 168 months with a mean of 52.8 months±49.6 SD. The most common manifestations were abscesses in 79.3% (deep organ abscesses in 37.9% of patients), followed by pneumonia in 75.8% and gastrointestinal symptoms in 27.5%. Rare but fatal complications were also reported among patients as one patient developed haemophagocytic lymphohistiocytosis (HLH) syndrome.
Although X linked-CGD universally constitutes the most common pattern of inheritance; only 6 of our patients 6/25 (24%) belonged to this group with a Stimulation Index (SI) of 1–5, and confirmed by carrier pattern of their mothers. Mothers were not available for testing in four male children. Nineteen patients (76%) had autosomal recessive patterns; ten males and nine females patients based on having abnormal SI, positive history of consanguinity and their mothers showing normal SI.
ConclusionIncreasing the awareness of physicians about symptoms of CGD may lead to earlier diagnosis of the disease, thus enhancing proper management and better quality of life.
Chronic granulomatous disease (CGD) is a primary immune deficiency disease that results from an inherited defect in the phagocytic cells of the immune system.1 The disease occurs due to mutations of the genes that encode the components of nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) which is responsible for transfer of electrons from NADPH in the cytosol across the phagosomal membrane to reduce oxygen to super oxide anion and deliver protons necessary for dismutation that produce hydrogen peroxide. The produced reactive oxygen intermediates (ROI) damage the phagocytosed microorganisms.2
The enzyme is formed of five subunits: gp91phox (CYBB) and p22phox (CYBA) which are integral membrane proteins that form the flavocytochrome b588 (the electron transport centre of the enzyme)3 and p47phox; neutrophil cytosolic factor 1(NCF1), p67phox; neutrophil cytosolic factor 2(NCF2), p40phox; neutrophil cytosolic factor4 (NCF4) which are cytosolic components.4,5
The gene encoding gp91 is found on the X chromosome, while other genes are located on autosomes.6 Thus inheritance can be X linked (about 70% of cases) or autosomal recessive (about 30% of cases).7
Autosomal forms are more reported in certain areas such as Turkey and Iran due to the high rate of consanguineous marriages in these countries.4
The prevalence of CGD varies from one in 1,000,000 to one in 160,000 individuals depending on the populations investigated and clinicians’ awareness.8 The incidence to be reported is expected to be higher among the Arab population (1:111,000).9
The clinical presentations of the patients are heterogeneous, however most of them present with development of severe and recurrent bacterial and fungal infections. Most of the patients suffer from pneumonia, lymphadenitis, hepatosplenomegaly and abscesses.1 However, diarrhoea and sepsis syndromes may also be a presenting feature and CGD may be misdiagnosed as Crohn's disease.7 They may also develop granulomas and premature death.1
Laboratory diagnosis of CGD depends on the inability of phagocytes from affected individuals to produce a normal respiratory burst, this can be measured by direct measurement of superoxide production, super oxide dependent ferricytochrome c reduction, nitroblue tetrazolium test (NBT) and oxidative test dihydrorhodamine (DHR) by flow cytometry, which is progressively replacing other tests due to its higher reproducibility, objectiveness, rapidness and ability to detect X linked carriers.1,5
There is a paucity of data on CGD from developing countries especially the Middle East with the high consanguinity, and speculated higher incidence than reported. This study aimed at describing the characteristics of Egyptian children with CGD.
Patients and methodsPatientsThe study was conducted in the Immunology Unit, Paediatric Department, Cairo University from 2012 through 2014. It was approved by the institutional review board and informed consents were obtained from children's guardians.
Among 117 patients screened for CGD, 29 patients were enrolled according to the following inclusion criteria suggestive of CGD (presentation with one or more of the following features): suppurative lymphadenitis, chronic diarrhoea, recurrent pneumonia, perirectal abscess and fistula, abscesses in the liver, spleen, lungs and/or brain, fungal infections, osteomyelitis, septicaemia and granulomas of the skin.
Patients underwent detailed clinical history taking and thorough examination with emphasis on their presenting symptoms, age at presentation, complications, outcome and detailed laboratory parameters.
Available family members were tested to determine the mode of inheritance whenever possible.
Diagnosis of CGD was based on flow cytometry Dihydrorhodmaine (DHR) 123 assay.10,11
One hundred age and sex matched healthy donors (attending for elective procedures e.g. tonsillectomy, circumcision) were included as a control group (mean age 60 months±15 SD), the mean fluorescence intensity (MFI) of the stimulated and non-stimulated granulocytes were determined to set the cut-off value for the stimulation index among the Egyptian population.
Dihydrorhodamine testPeripheral blood was obtained from patients and healthy controls, one hundred microlitres of whole EDTA blood was diluted 1:10 with phosphate buffered saline (PBS) (Lonza, Belgium) in two test tubes, then 0.5μl DHR 123(Sigma–Aldrich) at a concentration of 2.5μg/ml was added to each tube.
After incubation for 15-min, an enzyme stimulant; Phorbol 12-myristate 13-acetate (PMA) (Sigma–Aldrich) was added to one tube marked stimulated at a concentration of 160ng/ml and the tube was incubated for an additional 15min at 37°C in water bath.
At least 10,000 events were acquired on a CYTOMICS FC 500 Flow Cytometer (Beckman coulter, FL, USA).
The neutrophil stimulation index (SI) was calculated as the ratio of mean fluorescence intensity (MFI) of the stimulated cells to MFI in non-stimulated cells. The neutrophils were characterised by forward and side scatter properties.
In cases in which two distinct fluorescent populations were observed, MFI of each population was obtained and the SI for each population was calculated.
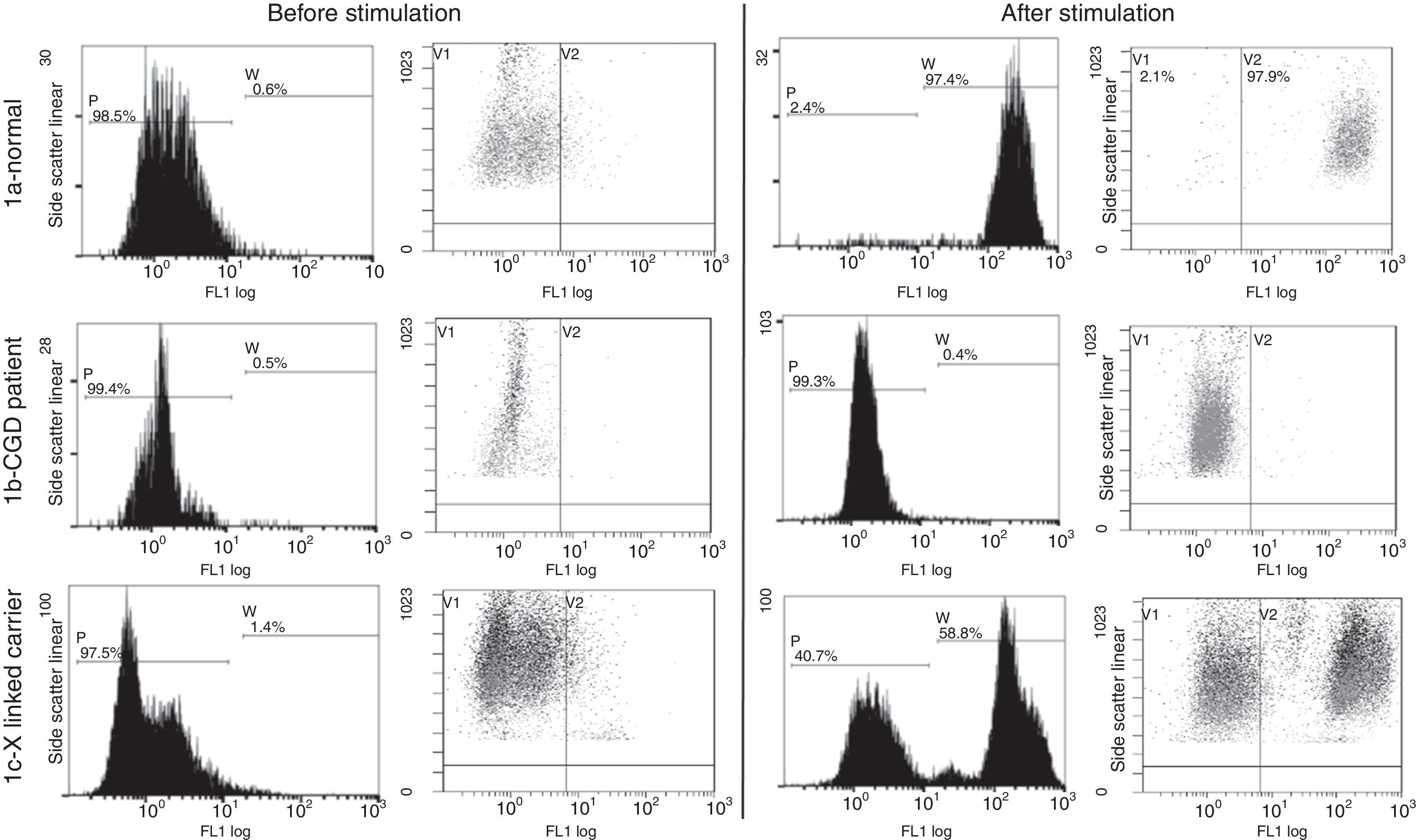
The flow cytometric analysis for 100 normal individuals was done (Fig. 1a) and the stimulation index of 70 was set as the cut-off value.
. (1a) Normal pattern in healthy controls: there is increase in the mean fluorescence intensity after stimulation of the neutrophils with phorbol myristate acetate. (1b) Patient no. 3 with disease pattern: there is minimal or no increase in the MFI after neutrophil stimulation. (1c) Mother of patient no. 3 with CGD carrier Pattern: there are two neutrophil populations, one normal and one affected.")
Flow cytometry dihydrorhodamine test done for functional assessment of NADPH enzyme function (gating on the neutrophils). (1a) Normal pattern in healthy controls: there is increase in the mean fluorescence intensity after stimulation of the neutrophils with phorbol myristate acetate. (1b) Patient no. 3 with disease pattern: there is minimal or no increase in the MFI after neutrophil stimulation. (1c) Mother of patient no. 3 with CGD carrier Pattern: there are two neutrophil populations, one normal and one affected.
Immunophenotyping of peripheral blood lymphocytes and immunoglobulins assay was performed for some patients to exclude other primary immunodeficiency disorders.
Serum immunoglobulin IgG, IgM, IgA were estimated by nephlometry. Complete blood count with the differential leucocytic count was done and lymphocyte subset were analysed using flow cytometry; blood was incubated with monoclonal antibodies to CD4 FITC, CD8PE, CD3 ECD, CD19 PE CY5, CD56 PE CY7 (Beckman Coulter, France) for 20min in the dark, then RBCs were lysed using Versa lyse (Beckman coulter), cells were washed, resuspended in FACS buffer and acquired on Cytomic FC500 flow cytometer, acquiring at least 10,000 events.
X-ray and CT scan were done when indicated to some patients.
Statistical methodsData were statistically described in terms of mean/standard deviation (SD) or frequencies (number of cases) and percentages when appropriate. Independent sample t test was used to compare between groups. All statistical calculations were done using the computer program SPSS (Chicago, IL, USA) version 15 for Microsoft windows.
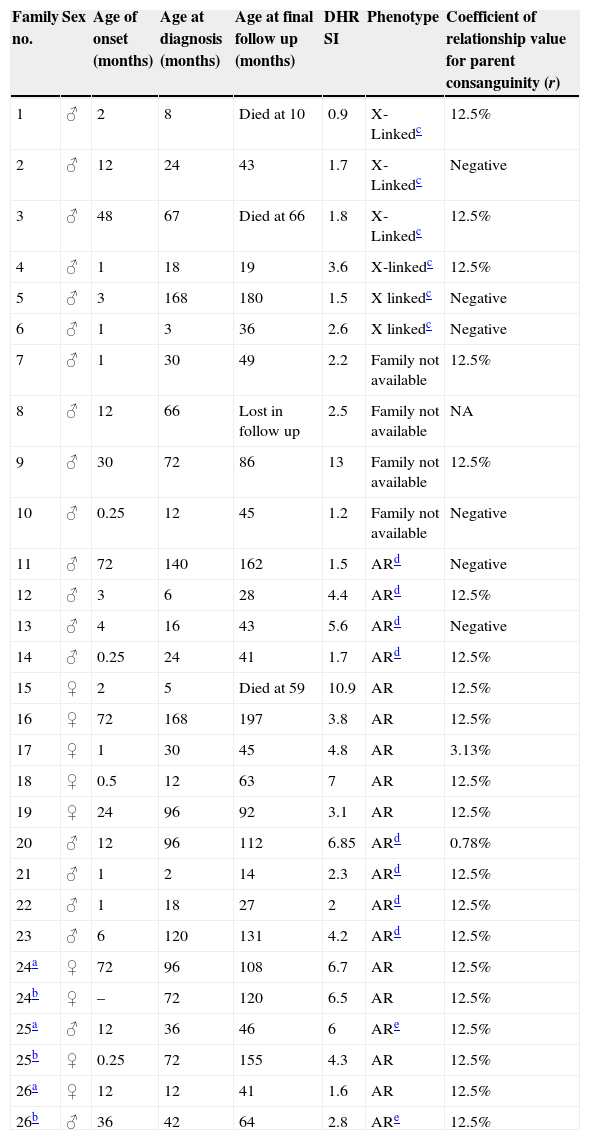
ResultsTwenty-nine patients (20 males and 9 females) from 26 different families are described (Table 1).
Descriptive data of the CGD patients.
| Family no. | Sex | Age of onset (months) | Age at diagnosis (months) | Age at final follow up (months) | DHR SI | Phenotype | Coefficient of relationship value for parent consanguinity (r) |
|---|---|---|---|---|---|---|---|
| 1 | ♂ | 2 | 8 | Died at 10 | 0.9 | X-Linkedc | 12.5% |
| 2 | ♂ | 12 | 24 | 43 | 1.7 | X-Linkedc | Negative |
| 3 | ♂ | 48 | 67 | Died at 66 | 1.8 | X-Linkedc | 12.5% |
| 4 | ♂ | 1 | 18 | 19 | 3.6 | X-linkedc | 12.5% |
| 5 | ♂ | 3 | 168 | 180 | 1.5 | X linkedc | Negative |
| 6 | ♂ | 1 | 3 | 36 | 2.6 | X linkedc | Negative |
| 7 | ♂ | 1 | 30 | 49 | 2.2 | Family not available | 12.5% |
| 8 | ♂ | 12 | 66 | Lost in follow up | 2.5 | Family not available | NA |
| 9 | ♂ | 30 | 72 | 86 | 13 | Family not available | 12.5% |
| 10 | ♂ | 0.25 | 12 | 45 | 1.2 | Family not available | Negative |
| 11 | ♂ | 72 | 140 | 162 | 1.5 | ARd | Negative |
| 12 | ♂ | 3 | 6 | 28 | 4.4 | ARd | 12.5% |
| 13 | ♂ | 4 | 16 | 43 | 5.6 | ARd | Negative |
| 14 | ♂ | 0.25 | 24 | 41 | 1.7 | ARd | 12.5% |
| 15 | ♀ | 2 | 5 | Died at 59 | 10.9 | AR | 12.5% |
| 16 | ♀ | 72 | 168 | 197 | 3.8 | AR | 12.5% |
| 17 | ♀ | 1 | 30 | 45 | 4.8 | AR | 3.13% |
| 18 | ♀ | 0.5 | 12 | 63 | 7 | AR | 12.5% |
| 19 | ♀ | 24 | 96 | 92 | 3.1 | AR | 12.5% |
| 20 | ♂ | 12 | 96 | 112 | 6.85 | ARd | 0.78% |
| 21 | ♂ | 1 | 2 | 14 | 2.3 | ARd | 12.5% |
| 22 | ♂ | 1 | 18 | 27 | 2 | ARd | 12.5% |
| 23 | ♂ | 6 | 120 | 131 | 4.2 | ARd | 12.5% |
| 24a | ♀ | 72 | 96 | 108 | 6.7 | AR | 12.5% |
| 24b | ♀ | – | 72 | 120 | 6.5 | AR | 12.5% |
| 25a | ♂ | 12 | 36 | 46 | 6 | ARe | 12.5% |
| 25b | ♀ | 0.25 | 72 | 155 | 4.3 | AR | 12.5% |
| 26a | ♀ | 12 | 12 | 41 | 1.6 | AR | 12.5% |
| 26b | ♂ | 36 | 42 | 64 | 2.8 | ARe | 12.5% |
AR: autosomal recessive, SI: stimulation index, NA: not available.
Among 117 patients investigated, 29 patients showed abnormal SI; 20 males (69%) and 9 females (31%). The consanguinity was 76% (19/25) among our patients; in the nine female patients, consanguinity was 100% (9/9) of cases. In male patients consanguinity was 68.4% (13/19). Thirty-two percent of the patients had history of sibling deaths. Forty-three percent of the patients were from Cairo, 36% from Delta cities and 21% from Upper Egypt.
The age at diagnosis ranged from 2 months to 14 years (mean: 52.8±49.6 month), while the mean age of first presentation was 15.7±23.1 months with 73% of the patients manifested in the 1st year with a mean diagnostic lag of 37 months.
Manifestations and infectious complicationsRecurrent skin and subcutaneous abscesses were the commonest clinical manifestations in 23/29 patients (79.3%), deep organ abscesses were detected in 11/29 patients (37.9%) (4 liver abscesses, 3 brain, 2 lung, 1 perianal and 1 splenic), followed by pneumonia in 22/29 patients (75.8%). Gastrointestinal symptoms in the form of diarrhoea, abdominal distention and pain were present in 8/29 patients (27.5%). Lymphadenitis and osteomyelitis were detected in 7/29 patients (24.1%); three patients with lymphadenitis had BCGosis secondary to BCG vaccination. Hepatosplenomagaly were detected in 6/29 patients (20.6%); four of them had liver abscesses and one had a splenic abscess. Draining ears were present in 6/29 patients (20.6%), less common manifestations like oral thrush were found in 3/29 patients (10.3%), neonatal sepsis in 1/29 (3.4%) and a rare but fatal complication haemophagocytic lymphohistiocytosis was reported in one patient (Fig. 2).
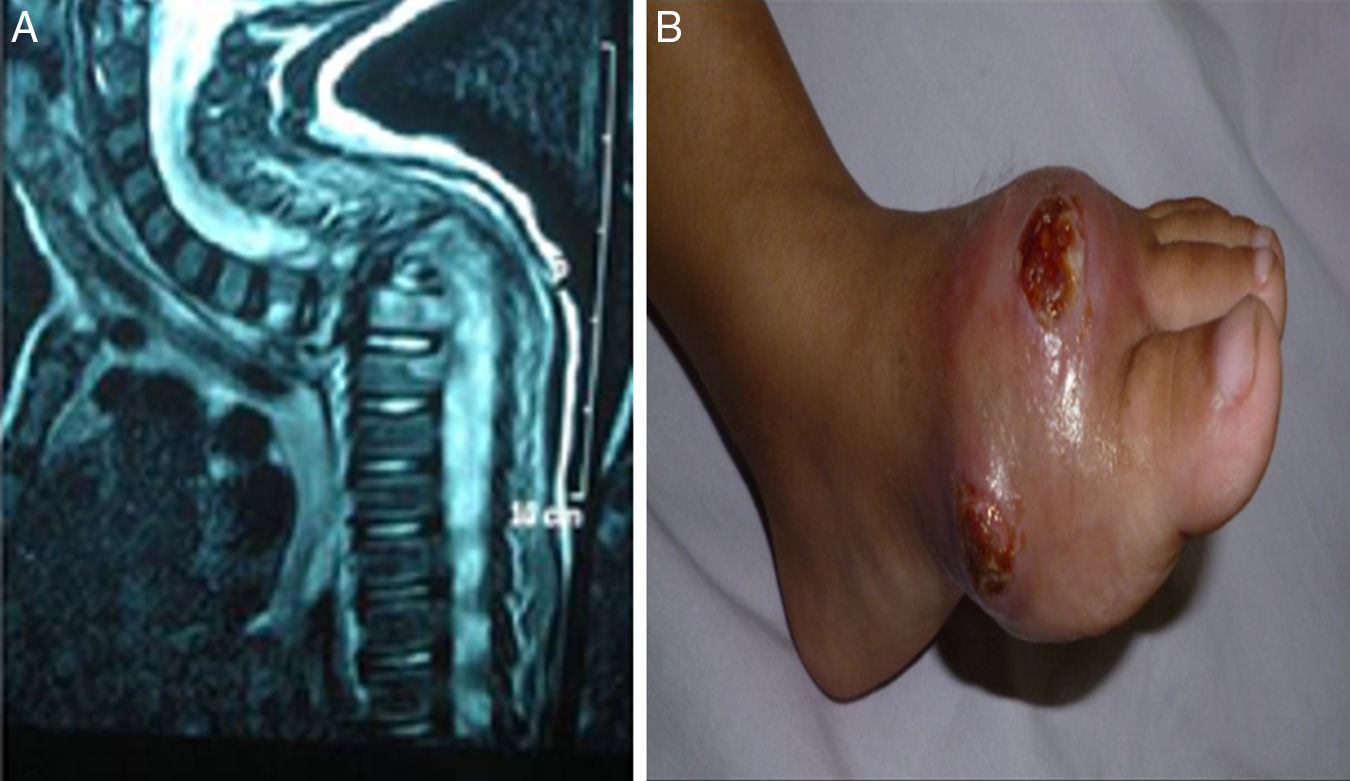
. (A) MRI cervical and spine with IV contrast showing marked kyphotic deformity, collapsed D2 and D3 vertebral bodies with consequent compression of the cord at that level (complication of HLH). (B) Osteomyelitis, ulceration with destruction of 1st metatarsal bone as complication of infection with atypical mycobacteria.")
(patient no. 26b). (A) MRI cervical and spine with IV contrast showing marked kyphotic deformity, collapsed D2 and D3 vertebral bodies with consequent compression of the cord at that level (complication of HLH). (B) Osteomyelitis, ulceration with destruction of 1st metatarsal bone as complication of infection with atypical mycobacteria.
Although localised brain abscesses were mentioned as an exceptional complication in CGD patients,17 we had three patients suffering this complication. One of those patients had history of Aspergillus pneumonia after being treated with Amphotericin, he developed seizures and manifestations of increased intracranial pressure. The pathology and microbiology of the excised mass showed hyphae of Aspergillus sp. fungi. The other two patients were referred after history of increased intracranial pressure manifestations, space occupying lesions by imaging in the parietal lobe and a provisional diagnosis of query malignant growths. This emphasizes that CGD should be considered in the differential diagnosis for all children presenting with brain abscesses and invasive fungal infections, particularly those involving the central nervous system.18
Eight patients had cultures results; the most common isolated infecting organism was Staphylococcus aureus (in 3/8 patients), Aspergillus sp. in 3/8 patients. Other infecting organisms were; Klebsiella, Pseudomonas aeruginosa, atypical mycobacteria strains, and Candida albicans (1 patient for each) (patient 20 had recurrent attacks of infections including Candida, Staph. aureus and Aspergillus).
All patients received oral antimicrobial prophylaxis in the form of Co-trimoxazole and Itraconazole. One patient received successful allogeneic fully-matched bone marrow transplantation. One patient received chemotherapy for the HLH. None of our patients had received therapy with IFN-γ because of limited resources.
The outcome of our cases was three deaths, two of them (patients no. 1 and 3) from a severe fulminant Aspergillus infections, both of them showed X-linked pattern. The third patient (no. 15) from severe chest infection.
Dihydrorhodamine assaySix of our 25 patients (24%) from six families belonged to the X-linked group with stimulation index ranging from 1–5 (Fig. 1b), the mothers of such patients were examined and they showed a carrier pattern with two peaks; one with stimulation index around one and the other with normal stimulation index above 70 (Fig. 1c). Mothers were not available for testing for the additional four male patients.
Nineteen patients showed patterns of autosomal recessive CGD, ten male patients were suggestive of autosomal recessive CGD based on having abnormal SI and positive family history of consanguinity and their mothers showed normal granulocyte functions or having affected female siblings.
In the nine female patients, consanguinity was 100% of cases; suggestive of autosomal recessive pattern and the DHR test showed a pattern closer to the X-linked CGD with SI<5 in 6/10.
There were no significant correlations between the SI and age at presentation or the severity of symptoms.
On comparing the AR and the X-linked groups there were no statistically significant differences regarding the age of onset (p value=0.474), age of diagnosis (p value=0.78) or regarding the unfavourable outcome (mortality) (p value=0.07). Regarding the mortality rate which was 33.3% in the X-linked group (2/6) and the mortality rate among AR group was 6% (1/18; as one from the 19 AR-CGD lost his follow up).
Immunophenotyping and immunoglobulins assayMost of the patients showed a normal range of lymphocyte subsets. However four patients had abnormal results, two of them had CD4 lymphopenia. [Patient no. 15; CD4 25% (absolute count 825, reference (ref.) level for age 1400–5100) and patient no. 23; CD4 20% (absolute count 358, ref. level for age 400–2100).] Two patients had absolute lymphopenia with decreased CD3 and CD19 absolute counts in spite of normal percentages.
[Patient no. 6 had 50.6% CD3 (absolute count 637 with ref. level 2300–6500) and 19% CD 19 (absolute count 239 with ref. level 600–3000) and patient no. 18 had 56.1% CD3 (absolute count 561 with ref. level 1400–8000 and 8.9% CD19 (absolute count 89 with ref. level 600–3100)].
Immunoglobulins levels were assessed for the patients, most of the patients had hypogammaglobulinaemia except two patients [patient no. 8 had IgG 408mg/dl (ref. level 584–1509), patient no. 15 had IgG 166 (ref. level 208–868) and IgM 30.4mg/dl (ref. level 32–120)].
DiscussionIn this single-centre study twenty-nine patients were diagnosed as CGD based on the abnormal SI of the DHR test and their clinical symptoms.
The number of patients recorded in this study is considerably higher than many other studies with relatively short reporting periods and single centre data.
The mean age at diagnosis was 52.58±49.6 months (4.25 years) which is older than that recorded in other studies such as Kobayashi et al. who included 23 patients where the mean age was 2.8 years ranging from 0.7 to 10 years. Another study by Soler-Palacin et al. reported that the mean age of diagnosis was 3 years.1,12 Whereas Rawat et al. and Baba et al. had their mean age at diagnosis as 4½ years and 5.1 years respectively.13,14 This raises the attention to a relative delay in the diagnosis of CGD in Egypt and the developing countries despite patients presenting early as the mean age of first presentation was at 15.7±23.1 months with 37 months lag. Geographical distributions of the patients denote lower number of patients from Delta Cities and Upper Egypt, despite the higher rate of consanguinity pointing to defective awareness in those areas.
Two patients from consanguineous families were diagnosed upon screening when their siblings were diagnosed as CGD. Their manifestations were minimal but positive results highlighted the importance of screening family members of affected patients. The X-CGD patients manifested very early in life, usually during the first year; their mean age was 11.1 months±18 SD, while the mean age of presentation in AR patients was 18.3 months±26SD. These results are in concordance with Köker et al.15 However, the mean age of diagnosis was 48 months±63 SD in X-CGD and 55.9 months±50SD in AR-CGD.
Similar to other studies such as those of Rawat et al. and Wolach et al. most patients had recurrent skin abscesses, pneumonia, deep organ abscesses.13,16 Although localised brain abscesses were mentioned as an exceptional complication in CGD patients,17 we had three patients suffering this complication.
Although X-linked patients account for 65–70% of all cases recorded in the western countries,19–21 in our study only six of our 29 patients (20.6%) from six families belonged to this group. These results are consistent with those found in Israel and India where the autosomal recessive pattern was 63% and 58%, respectively, because of the high consanguinity rates.13,16
For many of our autosomal recessive cases, we obtained a very low SI closer to the X-linked recorded pattern as in cases 10, 11, 14, 26a, 26b and 22. In those cases, study of the affected proteins and subsequent sequencing would be of value to evaluate whether the defective protein may affect the pattern of the DHR results.
Female carriers possess only one copy of the mutated gene on the X chromosome, and subsequent to lyonisation, have two populations of phagocytes: those which are gp91phox positive and those which are negative. Random X chromosome inactivation early in the foetal development of haematopoietic precursor cells may lead to two unequal populations.22 Regarding the immunophenotyping of the lymphocytes and the immunoglobulin levels most of the patients showed normal ranges of lymphocyte subsets except four patients; two showed CD4 lymphopenia which could be attributed to the existing infections in concordance with Rawat et al.13 Most patients had hypogammaglobulinaemia except for two patients; one with decreased level in IgG and IgA and the other with decreased level in IgG only. Similar observation was found by Hanoglu et al.23 and Keleş et al.24 as each described a patient with CGD and hypogammaglobulinaemia, another three reports of CGD and selective IgA deficiency were described. The cause of hypogammaglobulinaemia may be due to primary T and B cells immunodeficiency or secondary immunodeficiency caused by drugs, infectious diseases or other systemic conditions leading to hypercatabolism or excessive loss of immunoglobulins.23
The mortality among our patients was 3/28 (10.7%) (2 X-linked and 1 AR) which is lower than that reported by Soler-Palacin et al. and Baba et al. studies which was 31% and 33.3% respectively; this may be due to the long period of follow up 25 years1,14 and the Kobayashi study which was 13%.12 The three patients died because of severe chest infections, two of them with severe fungal infections (Aspergillus), this coincides with Köker et al.15 study that found the main cause of death to be Aspergillus infection in major organs such as the brain and lung.
ConclusionWe believe there are number of cases who are neither registered nor diagnosed as CGD or who have not been referred for immunological assessment. There is almost a lag of diagnosis in some cases in spite of the early presentation which is probably due to delayed referring to tertiary centres and under estimation of Primary immune deficiency as a possibility.
Increasing the awareness of physicians about symptoms of CGD may lead to earlier diagnosis of the disease, thus enhancing proper management and better quality of life.
Conflict of interestThere is no conflict of interests.
Ethical disclosuresProtection of human subjects and animals in researchThe authors declare that no experiments were performed on humans or animals for this investigation.
Patients’ data protectionThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article.
This work is partially funded by Cairo University.
