



INTRODUCTION
Parietaria judaica-induced pollinosis is an important clinical feature in the Mediterranean area1. The efficacy of subcutaneous injection of vaccines containing inhalant allergen is well established2. However, they can induce systemic reactions and require a tedious and long buildup phase. Because of the problems, ways of avoiding them have been investigated.
An approach to the modification of allergens that has been extensively studied is polymerization with the use of glutaraldehyde as a cross-linking agent. With this procedure, the allergen ability to react with human IgE antibody is reduced, retaining antigenic determinants accessible for the induction of IgG-class antibodies3-7. These glutaraldehyde-modified allergic vaccines are safer than unmodified vaccines, while retaining clinical efficacy6,7.
A method, including a depigmentation step in which the enzymatic activity is inactivated, pigments removed and the solubility of the allergoid enhanced, has recently been developed and used in several clinical and in vitro studies.
Pollen extracts, even after dialysis in 3 kDa membranes, retain irrelevant low molecular weight components which remain adsorbed to the allergenic proteins8. These substances consist of condensed flavonoids and/or catechins, which may interfere in the reaction of glutaraldehyde with protein amino groups. The depigmentation step removes these contaminants. The polymerization of depigmented pollen allergens leads to a second generation of allergoid vaccines with considerably lower IgE-binding potencies and higher solubility than observed with the first generation polymers based on non-depigmented starting materials.
The objective of this study was to investigate the safety and clinical efficacy of a chemically modified (depigmented and glutaraldehyde-polymerized) vaccine of Parietaria judaica. Based on the recommendations of the Nordic Guidelines, objective outcomes of efficacy such as specific nasal provocation tests and skin testing9 were considered, as well as subjective outcomes, such as symptom scores and visual scale.
MATERIAL AND METHODS
Study design
This clinical trial was conducted following Good Clinical Practices. The study was authorized by the Ethics Committee of the Hospital "Virgen de la Arrixaca", and the Spanish Health Authorities. All patients gave their witnessed written consent to participate in the study. The study was controlled, parallel and randomized, and included 2 groups of patients. One group treated with the modified allergen extract (group A). A second group (group C) was used as control, only receiving symptomatic pharmacologic medications.
Patient population
Patients were recruited from the outpatient clinic Allergy Department at the Hospital "Virgen de la Arrixaca" of Murcia, Spain. Thirty patients (10 male, 20 female) of a mean age of 31 years (range 19-40) were selected for the study. All patients met the following criteria: a clinical history of more that 2 years of evolution of rhinoconjunctivitis during the Parietaria judaica pollen season, positive skin-prick tests to a standardized P. judaica pollen extract (100 HEP/ml C.B.F. LETI, S.A.) and negative to the rest of common aeroallergens. A skin test was considered positive when the wheal size diameter was ≥3 mm in the absence of a reaction in the negative control. All patients had a positive specific IgE determination to Parietaria judaica pollen and a positive allergen-specific nasal provocation test (NCT). As exclusion criteria, we used those outlined in the WHO position paper on allergen immunotherapy2.
After the initial diagnostic tests, the patients were randomly assigned to one of the two groups. Group A received treatment with a maximum concentration of 2.4 μg of freeze dried modified allergen/ml, and group C pharmacologic treatment, consisting of antihistamines and occasionally oral corticosteroids.
Symptom and medication scores
During the pollen season, patients recorded daily symptom scores during the P. judaica pollen season. Chest (breathlessness, wheeze, chest tightness), nose (sneeze, blockage, and running), eye (itching, redness, streaming, and swelling), and mouth and throat (itching and dryness) were scored on a scale ranging from 0 to 3 (0 = none; 1 = slight, the symptom is clearly present but is not troublesome; 2 = moderate, the symptom is present, it is troublesome, but not disabling or insufferable; and 3 = severe, when the symptom is severe, disabling and/or insufferable). The daily total symptom score was calculated. The intake of medication was quantified according to Dreborg et al10. in which the total number of tablets and puffs in 24 hours made up the daily score.
Pollen counts
The pollen count was made accoding to a previously described technique11,12 with a standard Burkard volumetric spore-trap (Burkard Manufacturing Co., Rockmansword, Herst, U.K.). Pollen grains were counted daily (expressed as pollen grains/m3). The Parietaria juadica pollen season was defined as the period of 1997 having 90 % of the total yearly pollen grains, starting March 7th and ending August 12th.
Skin Prick Tests
Patients were skin tested on admission and after 12 months of treatment. The skin prick tests were conducted, in duplicate, on the volar surface of the forearm using extracts containing 100, 10, 1 and 0.1 HEP/ml (C.B.F. LETI, S.A., Spain). The same batch of native, unmodified allergen extract was used throughout the trial. The extract was supplied freeze-dried and vacuum closed to be reconstituted just before use. Histamine HCl 10 mg/ml and glycerinated saline solution were used as positive and negative controls, respectively. Skin tests were done between 9:00 A.M. and 11 A.M. None of the patients was pretreated with drugs, which could affect the performance of the test. Reactions were recorded after 15 minutes of application13. Wheal areas were outlined with a fine tip marker and transferred, with transparent tape, to the corresponding sheet of a Case Report Form. The area of each wheal was measured by planimetry using a Wacom pallette (Wacom Tehnology Co., USA) connected to the computer program MacDraft (Microspot USA, Inc.). For each patient, the individual bioequivalent dose of allergen extract to achieve a wheal of the same size as positive control (individual 10 HEP) was calculated9.
Nasal Provocation Tests
Nasal challenges were considered the main outcome to document clinical efficacy of the treatment9. The test was performed at baseline and after 12 months using native, standardised, unmodified allergen extracts. The degree of nasal obstruction was measured by means of the nasal inspiratory peak flow meter (NIPF)14,15. The same batch of native, unmodified allergen extract at 10, 1, 0.5 and 0.1 HEP/ml was used throughout the trial and was supplied freeze-dried and vacuum closed to be reconstituted just before use. All patients were tested between 8 AM and noon. None of the patients was pretreated with drugs that could affect the performance of the test. The results were expressed as the number of patients that were positive to each one of the concentrations used in the challenge.
Allergen vaccine
The modified allergen extract was supplied by C.B.F. LETI, S.A. Deffated pollen grains of P. judaica were extracted in PBS followed by followed by dialysis in 3 kDa flat-bed ultrafiltration membranes (Pellicon®, Millipore, Madrid). The depigmentation step consisted of a controlled acid treatment to remove the remaining proportion of adsorbed pigments. The resulting depigmented allergen preparations were used to be treated with glutaraldehyde16.
The determination of Par j 1 content was performed by means of ELISA using monoclonal antibodies and scanning densitometry17. The total allergenic potency18 was measured using a reverse phase specific IgE binding inhibition ELISA19. The IgG binding activity was measured by means of an ELISA specific IgG inhibition assay20.
The native extract was used for all in vivo tests (NPT and dose-response skin prick tests), and the polymer was used to treat patients during 12 months.
Immunotherapy schedule
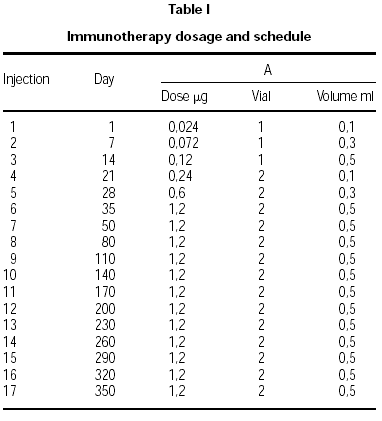
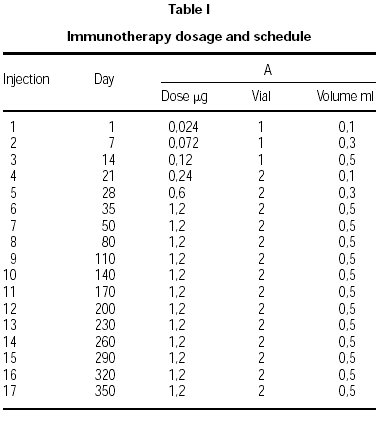
Two vials were prepared. Vial no. 1 was the result of depigmenting and polymerizing 10 HEP of the native allergen extract. Vial no. 2 contained a 10-fold higher concentration. The dose increment schedule is shown in table I. After maintenance was reached (5 weeks/6 injections), injections were given monthly. The indications for dose reductions were a systemic reaction (SR), large local reaction (ELR) defined as local swelling at the injection site 5 cm or greater in diameter 30 minutes after injection, or a late large local reaction (LLR) 10 cm or greater in diameter occurring 8 to 24 hours after injection. All the side reactions were recorded and were quantified according the EAACI guidelines21.
Statistics
The sample size of this study was based on previous observations for determining efficacy based on nasal challenges22. The calculation was based on a two-tailed, mean differences, with a criterion for significance (α) of 0.05, a power of 0.95, and assuming 20 % of abandons. Based on the previous characteristics, the study needed 15 cases per cell for a total of 30 cases. The software Systat (SPSS, Inc. USA) was used for statistical analysis. In addition to descriptive, a log-log regression in the bioassay of dose-response of in vivo skin prick tests was conducted23. The dose-response relationship between the geometric mean of the wheal and the concentrations used was calculated for each patient using the least squares method. The regression line analysis log (Y) = a + b x log (X), in which Y is the wheal area and X is the allergen concentration, was used23 to calculate the individual amount of allergen needed to achieve a skin reaction of the same size as histamine 10 mg/ml (individual 10 HEP). Daily symptom and medication scores (area under the curve AUC) during the pollination period24,25, and the individual 10 HEP were compared groupwise by the non-parametric test of Mann-Whitney.
The non-parametric Wilcoxon test was used to compare, into each group, the individual 10 HEP before the use of immunotherapy and after 12 months. The contingency table analysis was used to statistically evaluate the number of patients that were positive to each concentration of the allergen in each group at the beginning and at the end of the study.
RESULTS
Characteristics of the extract used
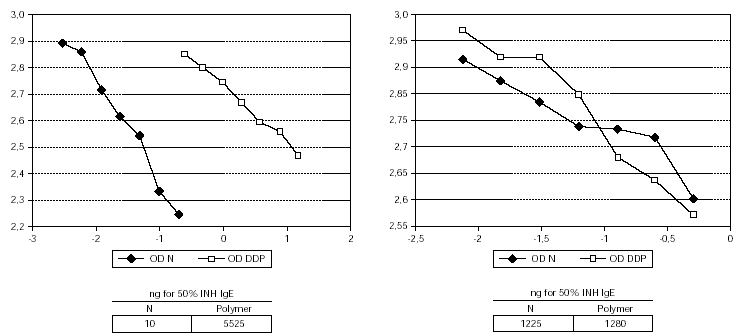
The 50 % inhibition of IgE binding was 10 ng for the native unmodified extract and 5525 ng for the resulting polymer (fig. 1). The potency for human IgG-antibody binding (50 % inhibition of specific IgG binding) of the native allergen extract and the polymer remained without modification (1.25 μg vs. 1.28 μg) (fig. 1). The native extract contained 363 μg of Par j per mg of freeze-dried extract and the polymer 12 μg.
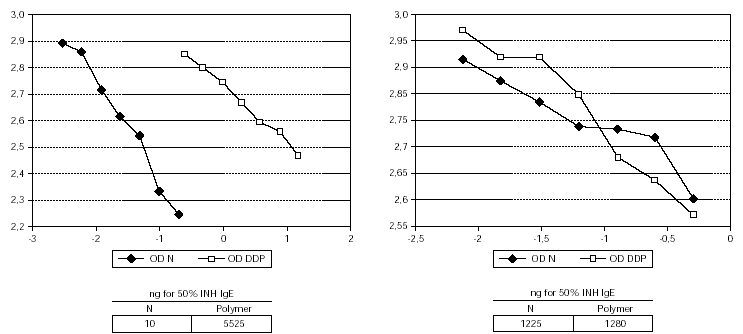
Figure 1.--Characteristics of the IgE and IgG binding inhibition of the native unmodified extract and the depigmented and glutaraldehyde polymerized extract. The modified extract needs a higher amount than native to achieve the same degree of IgE binding inhibition, whereas the IgG binding remains without modification.
Immunotherapy schedule and side effects
All the 30 patients finished the study and patients treated with Depigoid® reached the maximum dose. Systemic reactions and local reactions with a diameter over 5 cm were not reported.
Pollen count, symptom and medication scores
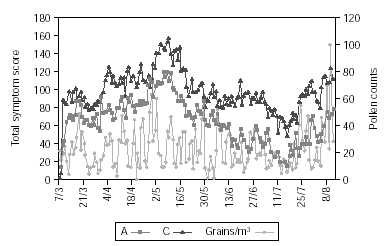
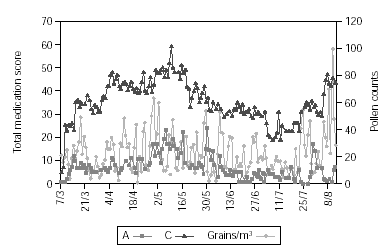
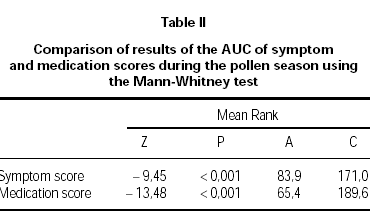
Figure 2 shows pollen counts and symptom scores during the pollen season and figure 3 the pollen counts and the medication score. Table II shows the statistical analysis (Mann-Whitney) of the effects of IT on symptoms and medication use between the groups. The differences are statistically significant (p < 0.001), being both scores lower in group A.
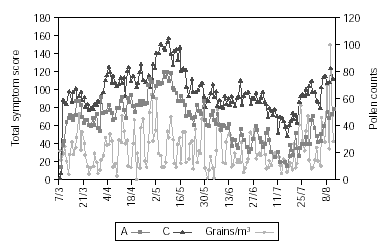
Figure 2.--The daily pollen count in the observation period and the daily group symptom score.
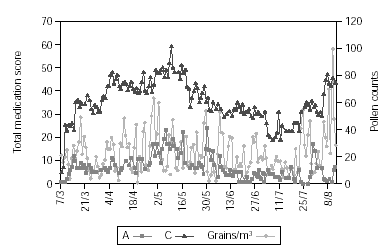
Figure 3.--The daily pollen count in the observation period and the daily group medication score.
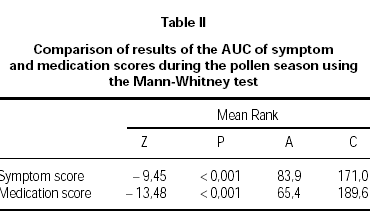
Table II Comparison of results of the AUC of symptom and medication scores during the pollen season using the Mann-Whitney test
Nasal provocation tests
Table III shows the number of patients of each group that had a positive NPT at baseline and after 12 months of treatment. The contingency table analysis shows that at baseline the nasal sensitivity between the two groups is not significant (Chi square: 4.3; p = 0.6), whereas, after 12 months of treatment only the group treated with the modified allergen extract shows an improvement that is significant (Chi square: 10.9; p = 0.004). The same analysis was applied to the number of patients in each group, who experienced an increase of the quantity of native allergen needed to achieve a positive nasal challenge. The results show that only group A has a significant clinical evolution (Chi square: 6.6; p = 0.037), whereas the control group remains without modification (Chi square: 4.8; p = 0.907).

Skin prick tests
The results are expressed as the amount of native, unmodified allergen extract necessary to induce a wheal of the same size as the positive control. At baseline, group A needed an average of 2.6 (IC 95 % 0.4-4,9), and group C 9.9 (IC 95 % 1.8-18). After 12 months of treatment, the results was 3 (IC 95 % 0,476-5,695) for group A, and 11,815 (IC 95 % 0,768-24,397) for group C. The statistical analysis shows that there differences between the two groups at baseline and after 12 months of treatment are not significant (p > 0.05).
When comparing skin test reactivity at baseline and after 12 months of treatment, both groups remained without significant changes.
DISCUSSION
This clinical trial was designed to evaluate safety and efficacy of a depigmented, glutaraldehyde modified P. judaica allergen extract. In terms of safety, there were no systemic reactions and only two local reactions with a diameter lower than 5 cm were recorded.
The recommendations outlined in the Nordic Guidelines were followed to demonstrate efficacy. These guidelines suggest that in cases with a known natural history of allergic respiratory diseases, open studies with a restricted number of patients are acceptable if a clinical effect of the treatment is documented using objective methods, such as provocation tests, and the sample size is sufficient for statistical analysis of the results9. The evaluation of the specific sensitivity of the shock organ, before and after treatment, is the most objective parameter to analyze the efficacy of immunotherapy26. Thus, we used the nasal provocation test as an objective method to document the clinical effect and to calculate the sample size. We selected the nasal inspiratory peak flow (NIPF) as the method of choice to assess the nasal patency14. This method has a good correlation with anterior rhinomanometry15,27. The medication score and the subjective outcome of symptom score were significantly lower in the group treated with IT than in the C group (p < 0.01).
An important conclusion of this study is that the depigmented polymerized extract of Parietaria judaica protects against allergen present in the native extract. This suggests that clinically relevant epitopes are present in the polymer. These finding is in agreement with specific IgG data, which demonstrated that both, native and polymer, retain IgG binding epitopes, while IgE binding epitopes are drastically reduced in the polymer.
Glutaraldehyde-modified allergens have been shown to be able to modify cytokine production toward and favours IFN-gamma secretion with a subsequent change in the balance Th1 and Th2 activity towards a Th1 response and downregulation of IgE antibody28-30.
This study confirms that depigmented polymerized extracts of the pollen of Parietaria judaica are safe and effective in the treatment of Parietaria pollen allergic patients, and provide clinical benefit in the shock organ after 12 months of treatment. Symptom and medication scores were also improved. Depigmented polymerized extracts of the pollen of Parietaria judaica induce clinical protection against a native extract as verified by specific nasal provocation.
