




Common variable immunodeficiency (CVID) is classified as a predominantly antibody deficiency 1 (table I). It comprises a heterogeneous group of alterations all characterized by deficient antibody synthesis 2-4. In the past it was known as late-onset hypogammaglobulinemia, and earlier still was referred to as Giedion-Scheidigger deficiency or dysgammaglobulinemia due to the multiple combinations of immunoglobulin levels involved. CVID is related to selective IgA deficiency, and both abnormalities may often coincide in one same family 3. CVID can manifest at any age as recurrent bacterial infections, and is characterized by the presence of hypogammaglobulinemia with failure in the production of antibodies in response to different antigens. The number of B and T lymphocytes tends to be normal or almost normal, though important reductions in cell count are sometimes observed 5. The incidence of CVID ranges from 1/25,000 to 1/66,000 inhabitants, though the more milder cases probably go undetected 6. Although selective IgA deficiency is much more common, it is also frequently asymptomatic; consequently, CVID is considered to be the most frequent immune deficiency requiring patient treatment.

PATHOGENESIS
The defect underlying CVID is located in the terminal maturation phase of the B lymphocytes, affecting the production of antibody-generating plasma cells or the immunoglobulin class switch from IgM to IgG. The effect is generally intrinsic to the B cell population, though in some cases regulatory T cell function fails, with or without primary B cell deficiency. IL-2, IL-4, IL-5 and IFN-γ deficiency may be associated, and in some cases a CD40 ligand (CD40L) defect is observed though this appears to constitute a secondary alteration. Genetic and molecular studies have shown the coincidence in one same family, and even within one same individual, of cases of CVID and of selective IgA deficiency. It is believed that the carriers of certain mutations, depending on exogenous factors or complementary genes, develop isolated IgA deficiency in some instances and CVID in others, with different intensities and at different times even in adults. Thus, some of these families present mutations in genes of the HLA-III system, e.g., C2 and C4 factors, or TNF.
Immune alterations in CVID
Patients with CVID usually present hypogammaglobulinemia 7, and IgG and IgA are more affected than IgM, though there are multiple possible levels and combinations. It should be pointed out that immunoglobulin normality does not rule out CVID, and the definitive diagnosis requires confirmation of the lack of specific antibody response following protein and/or polysaccharide antigen challenge 8-10.
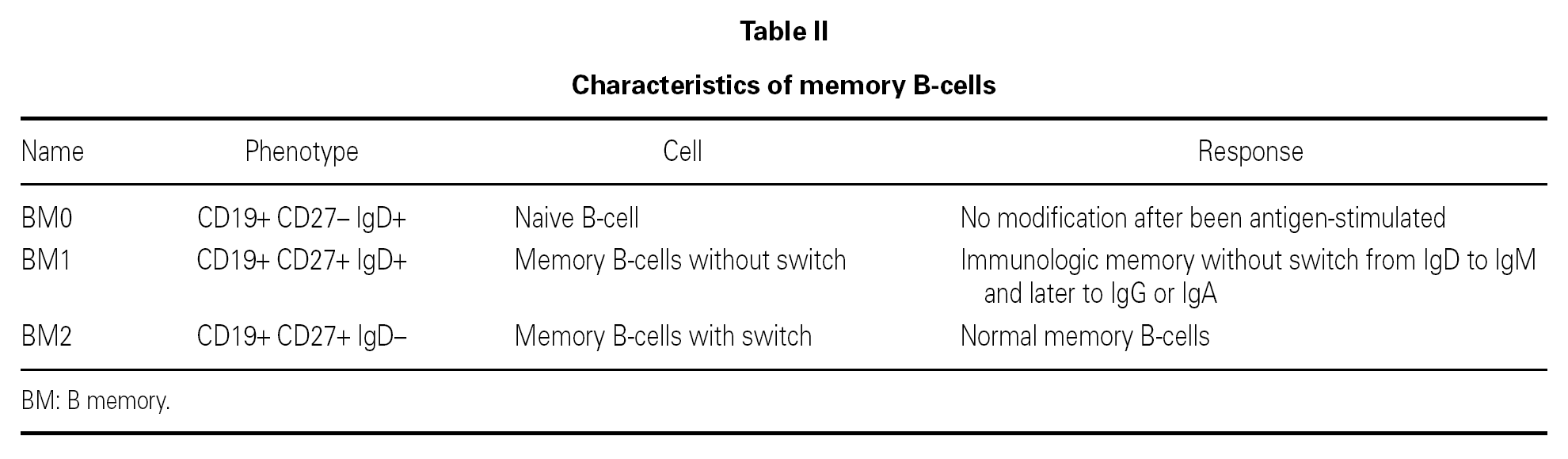
The B lymphocyte count is usually normal or almost normal, with a mature B phenotype, though in contrast the plasma cells of the lymphoid tissues are diminished in number. Nevertheless, imbalances in some B cell subpopulations have been found, such as the immature forms 11, and such populational anomalies may increase with patient age 12. The most relevant observation has been the detection of anomalies in the memory B cells, which serves to classify the different forms of CVID and to predict the course of the disorder in each patient 13-16 (table II). The reduction in memory B cells (CD19 + CD27 + IgD) is associated in both children and in adults to severe forms, with bronchiectasis and/or splenomegalia 17, though not so the immunoglobulin levels, which lack prognostic value 18. In contrast to what was expected, the situation in terms of the memory B lymphocytes was not seen to correlate to the genetic mutations recently described in CVID 18.
The T cells are seen to be normal in some patients, though other affected individuals present anomalies in proliferation or cytokine synthesis in response to different stimuli. T-B lymphocyte cooperation is particularly affected 7. Patients with serious complications tend to present a low CD4/CD8 ratio due to an increase in activated CD8 + lymphocytes (CD8 + HLA-DR +) 19. High counts of large granular lymphocytes (LGL) have also been reported 20.
Recently new anomalies have been described in CVID, though their relationship to the pathogenesis and clinical severity of the disease remains the subject of research, since they appear to manifest in some but not in all patients. These anomalies include innate immune defects, particularly in relation to the activation, development and function of the dendritic cells of monocytic origin 21,22. In some cases the defect is accompanied by variable alterations in the production of IL-12 23,24, which causes secondary anomalies in T cell activation, though no significant Th2 > Th1 predominance has been demonstrated 25. A defect in IL-7 synthesis has also recently been published that appears to be relevant, since it occurred in a subgroup of patients with CVID complicated by splenomegalia, autoimmune disorders and an increase in circulating CD8 + lymphocytes 26. Another recently identified failure in native immunity involves the TLR9 (toll-like receptor 9), which recognizes the CpG motifs present in viruses and bacteria a situation that could have defensive consequences 27.
CLINICAL MANIFESTATIONS
Although CVID is attributable to a genetic defect with immune failures that are present from birth, the clinical manifestations of the disease often only appear in adulthood though there have been reports of complications in patients aged 2 to 66 years 28. Of note is the variety of symptoms and their severity, which can be seen in members of one same family presenting the same mutation. The clinical manifestations generally begin in the form of bacterial respiratory infections, complicated years later by lymphoid hyperplasia, autoimmune processes, lymphomas or granulomas 2. Since the infections may not appear or may be of scant intensity, it is not unusual for the diagnosis of CVID to be delayed for years, until the complications appear.
Infections
Although the infections tend to manifest in adults, children may also be affected, with two peaks in frequency: one in the 1-5 years age range, and the other in the 16-20 years age interval 7. The most common clinical presentation consists of recurrent sinus-bronchial infections. At the time of diagnosis of the disease, most patients have already suffered some episode of bacterial pneumonia 5. The most frequently isolated pathogens are Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis and different staphylococci. It is also possible to find Pneumocystis jirovici (previously carinii), Mycoplasma pneumoniae and certain mycobacteria and fungi 6.
Late complications
Some patients, either before or after the recurrent respiratory infections, develop gastrointestinal problems, granulomas, autoimmune manifestations, lymphomas, or cancer. These complications are inherent to adults, but occasionally may also be found in children.
Chronic lung disease
Chronic lung pathology is very common, and many adults ultimately develop bronchiectasis despite adequate management from childhood 29. The risk of lung damage is associated to a deficient production of antibodies against bacterial polysaccharides 30, and to a decrease in memory B lymphocytes 16. Another common cause of chronic lung disease in adults with CVID is lymphocytic interstitial granulomatosis, which associates progressive dyspnea and is an indicator of poor prognosis, since it is usually accompanied by lymphoproliferative processes 31.
Granulomatosis
The etiology underlying granulomatosis is not clear, though it has been associated with a chronic infection due to human herpes virus 8 (VHH8) 32. Although the lungs are the most commonly affected region, granulomas may also appear in the skin, intestine or liver. Alternatively, generalized multisystemic presentations simulating sarcoidosis can be seen 33-35. Granulomatosis is an unfavorable finding, due to the treatment difficulties involved and its frequent association to autoimmune and lymphoproliferative processes 33,34.
Gastrointestinal manifestations
Some patients with CVID develop inflammatory bowel disease, Crohn's disease or ulcerative colitis in early or later stages 36. Although the clinical picture and histological findings may be typical, it is more common to observe atypical forms of inflammation with malabsorption, diarrhea and weight loss 37. Other possible clinical conditions are chronic malabsorption with steatorrhea and vitamin B12 deficiency; protein-losing enteropathy; lactose intolerance; and villous atrophy more often related to Giardia lamblia parasitosis than to gluten 5. Some cases of colitis have been associated to viral infection, the recommendation being to search for herpes virus or cytomegalovirus in CVID patients with colitis 38. Lymphoid hyperplasia, symptomatic or otherwise, is often identified if radiological explorations are carried out.
The risk of gastrointestinal infections is high in some patients with CVID the main causal agents being Salmonella, Shigella and Campylobacter. It has been reported that Helicobacter pylori infection occurs in 80 % of patients with CVID who suffer dyspepsia. Systematic evaluation of such infection is recommended, with eradication in view of the high risk of gastric cancer involved 39,40.
Rheumatological and autoimmune diseases
Approximately 20-25 % of all adults with CVID ultimately develop some autoimmune disorder, or a combination of several such disorders 41. These complications generally comprise rheumatological problems such as chronic arthritis, scleroderma, dermatomyositis, lupus erythematosus, and particularly Sjögren's syndrome 42. Other common problems include autoimmune cytopenias (hemolytic anemia, thrombopenia, neutropenia), and disorders such as hepatitis, biliary cirrhosis, Guillain-Barré syndrome, parotiditis, pernicious anemia, growth hormone deficiency, etc. Globally, these disorders are all more frequent in CVID than in selective IgA deficiency or in IgG subclass deficiency 43. In children, thrombopenic purpura is possibly the most common autoimmune disorder, and it should be pointed out that the hematological diagnosis often precedes that of CVID. Consequently, an immune evaluation is essential in the event of any atypical thrombopenia 44,45. CVID has been associated to insulin-dependent diabetes in children and adolescents 46,47, and a celiac patient with the typical DQ2 A1 0501 haplotype has been documented 48.
Cancer and lymphomas
Elderly adults with CVID have a high cancer risk-lymphomas and intestinal lymphoreticular processes being the most common disorders 5,49. Patients diagnosed with non-Hodgkin lymphoma may possibly present occult CVID 50. Extranodular marginal zone B lymphomas, previously known as MALT (Mucosa-Associated Lymphoid Tissue) lymphomas, are the most typical presentations. In contrast to the lymphomas of other immune deficiencies, these tend to be well differentiated, secreting immunoglobulins, and are characteristically negative for Epstein Barr virus 5. Gastric lymphomas have been associated with Helicobacter pylori, disappearing after triple antibiotic treatment. As a result, some authors recommend such treatment in all CVID patients with dyspepsia, even if the infection has not been demonstrated 50. The diagnosis of lymphoma is a particularly delicate matter, since the patients usually present lymphoid hypertrophy and benign adenopathies for many preceding years 25.
Lymphoproliferative infiltration is frequent, causing lymphoid hyperplasia in the form of adenopathies and splenomegalia, though infiltration of other organs is also observed, such as the liver or kidneys resulting in functional failure. The alterations are polyclonal, though malignization may occur. Their relation to B lymphomas, and the lymphoid lineage involved, is not clear. Recently, in a case of CVID with TACI mutation, the lymphocytes of the infiltration were identified as corresponding to T CD8 + cells 51.
DIAGNOSIS OF CVID
In view of the clinical variability of the disease and the limited usefulness of the genetic studies, the diagnosis of CVID is based on the immune findings. However, due to the heterogeneity of the disorder, no single protocol has been established, and adaptations to each individual case are required 4,5. Hypogammaglobulinemia is the most suggestive finding, though normal immunoglobulin levels do not rule out the diagnosis. Consequently, in suspect cases, evaluation is required of antibodies targeted to thymus-independent polysaccharide antigens or thymus-dependent protein antigens, e.g., vaccinating against pneumococcus and tetanus. Isohemagglutinins tend to be absent or present at low levels. Other studies of B and T cell population and subpopulation function or number are useful for defining the prognosis and risk of complications 13,16 (table III).

Differential diagnosis
The diagnosis of CVID is largely based on the exclusion of other immune deficiencies, though this is not always easy, since the disease shares many characteristics with other disorders. Some patients diagnosed with CVID afterwards have been shown to present Btk gene mutations the disorder actually corresponding to mild forms of sex-linked agammaglobulinemia 52. Differentiation from hyper-IgM syndrome based only on immune studies is a delicate matter, since IgM is not always increased, and because some cases of CVID show poor expression of the CD40L molecule despite no mutation of its encoding gene 52. The differential diagnosis with respect to chronic granulomatosis may prove difficult in some concrete cases, though a clue is provided by the older age of patients with CVID. The greatest differential diagnostic difficulty refers to selective deficiency of IgA, since its genetic and pathogenic relationship to CVID has been demonstrated, and a given patient may evolve from one disorder to the other 53. The differential diagnosis will become easier once more genetic information on CVID becomes available. For the time being, high IgM levels or a B lymphocyte population < < 2 % are immune data against a diagnosis of CVID 52.
TREATMENT
Years ago, cimetidine was evaluated in patients with CVID, though the results were disappointing. Posteriorly, pegylated IL-2 was administered 54,55. At present, IgG is considered the treatment of choice, and drastically reduces the incidence of respiratory infections 56,57. In the past, the treatment was started when the infections appeared, though IgG is known to prevent the pulmonary complications 57,58; consequently, it should be administered to all CVID patients with hypogammaglobulinemia, until serum IgG stabilizes at between 500-700 mg/dl. This requires the infusion of individualized doses of between 270-500 mg/kg/month 56,58. The administration of subcutaneous IgG on a rapid (20 ml/h) and domiciliary basis is increasingly popular in children and adults, because it is well tolerated, avoids hospital dependency and improves patient quality of life ensuring protection against infections similar to that afforded by administration via the intravenous route 59-61.
The rheumatic manifestations (Sjögren's syndrome and rheumatoid arthritis) improve by adding IgG to conventional therapy 62, though not so the cutaneous granulomas. Indeed, it is better not to treat the latter as long as they remain asymptomatic, because they tend to recur after surgical removal 34. Recently, remissions have been reported with anti-TNF (etanercept, infliximab) thus opening up new therapeutic perspectives for granulomatosis 63,64.
Management of the autoimmune cytopenias involves the usual treatment protocol. Good experience has been gained in relation to thrombopenias treated by splenectomy though logically the anti-infectious coverage must be extremely rigorous 28.
Antibiotic treatment should adhere to the cautions of choice, duration and rotation recommended for chronic infections. Some authors recommend the use of macrolides, because they offer a certain antiinflammatory capacity in addition to antibacterial action 65.
GENETIC AND MOLECULAR FINDINGS IN CVID
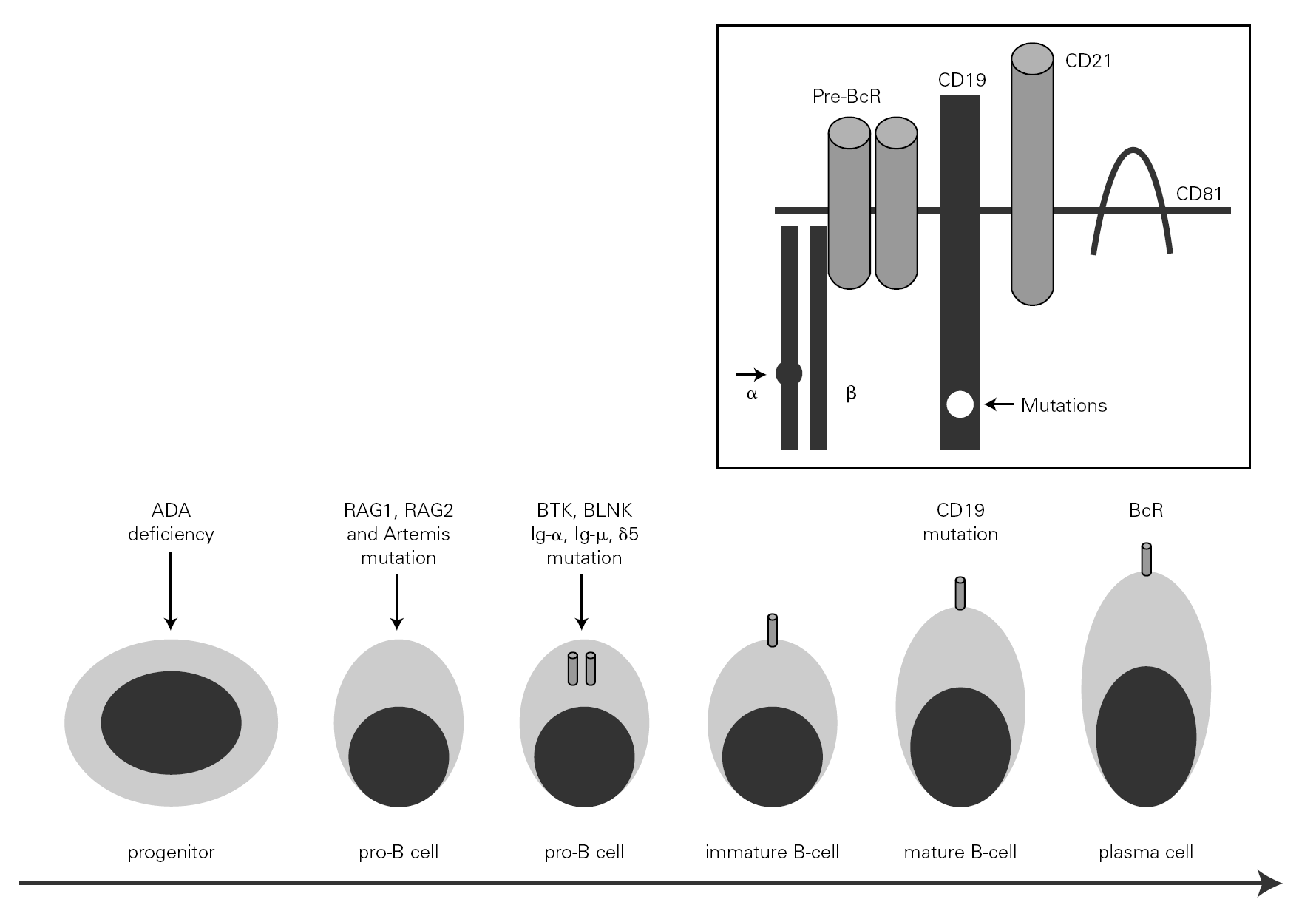
The mechanism underlying CVID remains unclear, and is certainly not the same for all forms of the disease. The theory popular during the eighties that CVID is an acquired disorder secondary to viral infection has been abandoned 66. Paradoxically, however, the correction of immune anomalies has been reported in CVID patients following infection with the human immunodeficiency virus 67. At present, CVID is considered to be a primary genetic alteration with a molecular mechanism that directly or indirectly affects B cell maturation and immunoglobulin synthesis (fig. 1).
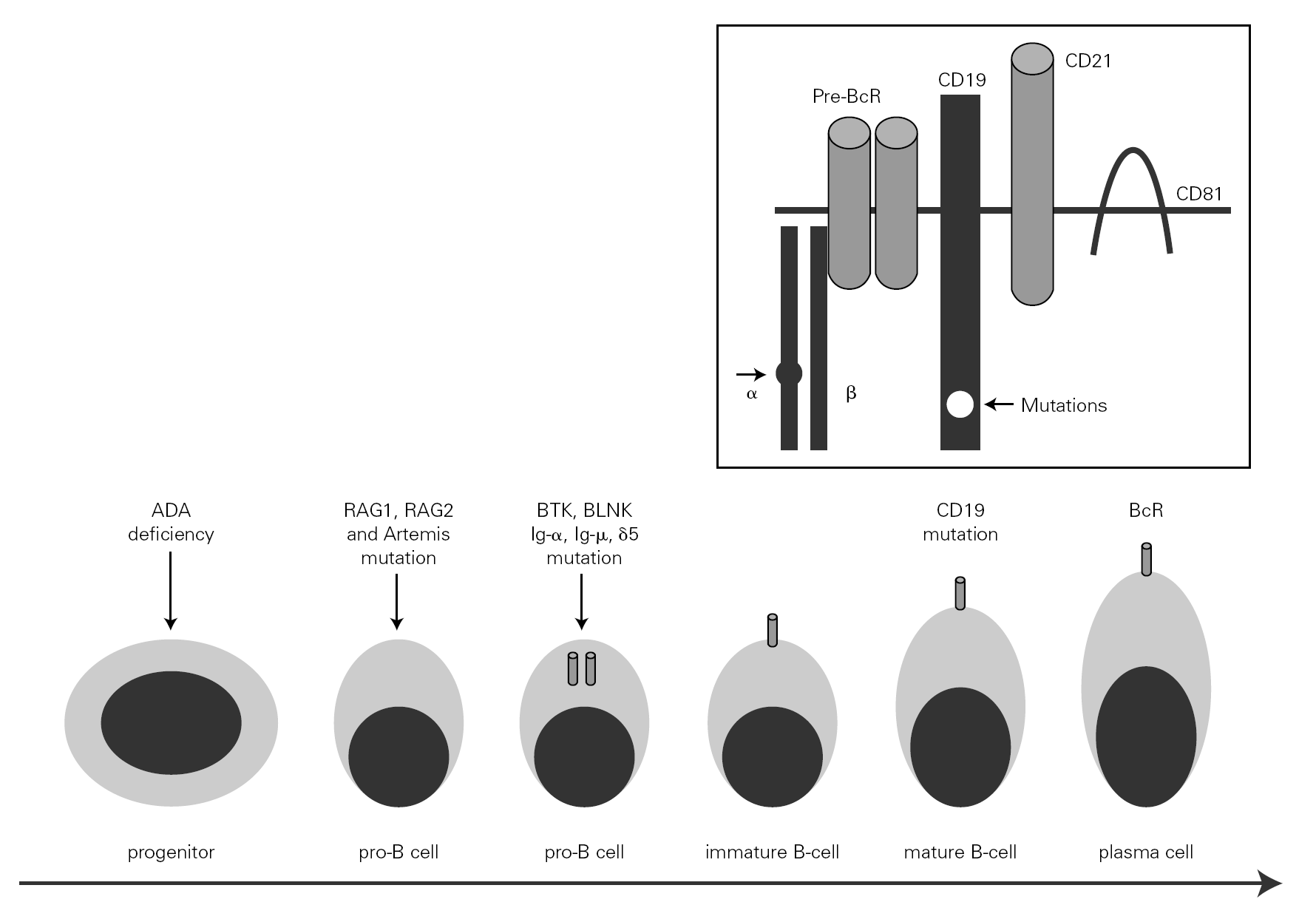
Figure 1.--B-cell differentiation from a progenitor stem cell to a pro-B, to a pre-B, and finally to a mature B-lymphocyte (some steps are not shown). The arrows indicate the B cell stages affected by genetic mutations causing immunodeficiency. Within the frame is represented the B cell receptor complex, with the two presently reported mutations, which are located in CD19 and the α chain. ADA affects very immature cells, producing relevant deficiencies; in contrast, CD19 or BcR defects occur in mature B cells. ADA: adenosine deaminase: RAG: recombinant-activating gene; BTK: Bruton's tyrosine kinase; BLNK: mutated B cell-linked protein.
Maturation of B lymphocytes immunoglobulin isotype switch
Two simultaneous processes are involved in the maturation of B lymphocytes: maturation of the cells to form plasma cells, and a switch in the immunoglobulin isotype synthesized, from IgD to IgM, and then to IgG or IgA without changing the specificity of the antibody. A detailed review of lymphocyte development has recently been published 68.
This switch, or more specifically CSR (class-switch recombination) takes place through DNA recombination and excision, and depends on expression of the AID (activation-induced deaminase) gene. This complex genetic process has drawn special attention 69. Its initiation requires two signals. The first signal comprises a release of cytokines involved in B cell maturation and in the synthesis of antibodies. Thus, TGFb activates the IgA heavy chain promoter, while IL-4 and IL-13 do the same for IgG and IgE. The second signal comprises intimate contact with other cells. For years cooperation with T lymphocytes has been known through the CD40 molecule of B lymphocytes and the CD40 ligand (CD40L) of the T lymphocytes, which activate the AID promoter in the same way as TLR9 (toll-like receptor 9) 70.
BAFF/APRIL system
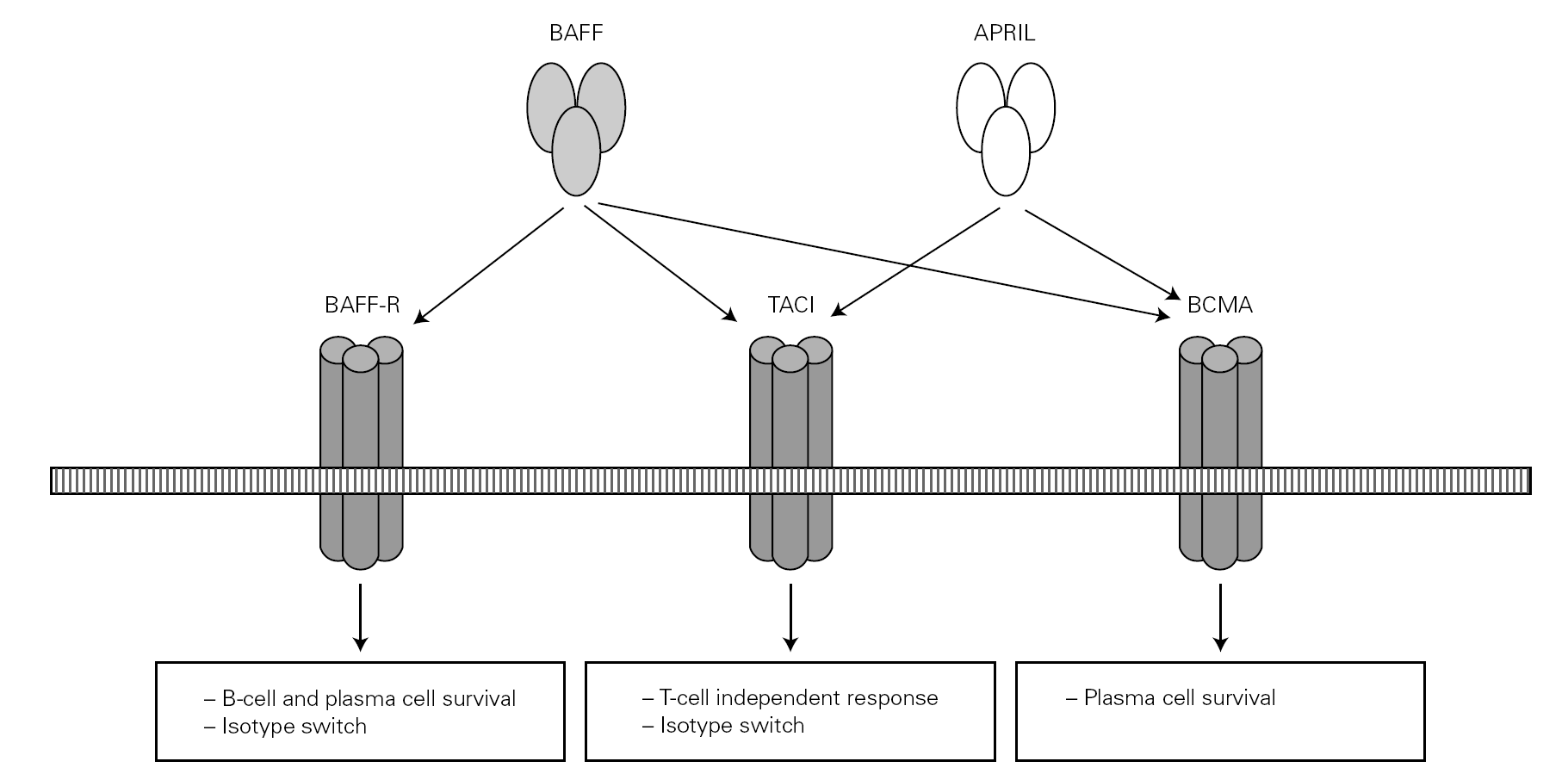
Posteriorly, a new cell cooperation system independent of the lymphocytes was discovered 71. This system is based on two membrane molecules of the TNF family (BAFF: B cell activating factor and APRIL: proliferation-inducing ligand). This mechanism allows the switch to IgG and IgA in mice previously subjected to CD40 + lymphocyte depletion thus demonstrating its independence of the CD40-CD40L lymphocyte route 71-73 (fig. 2).
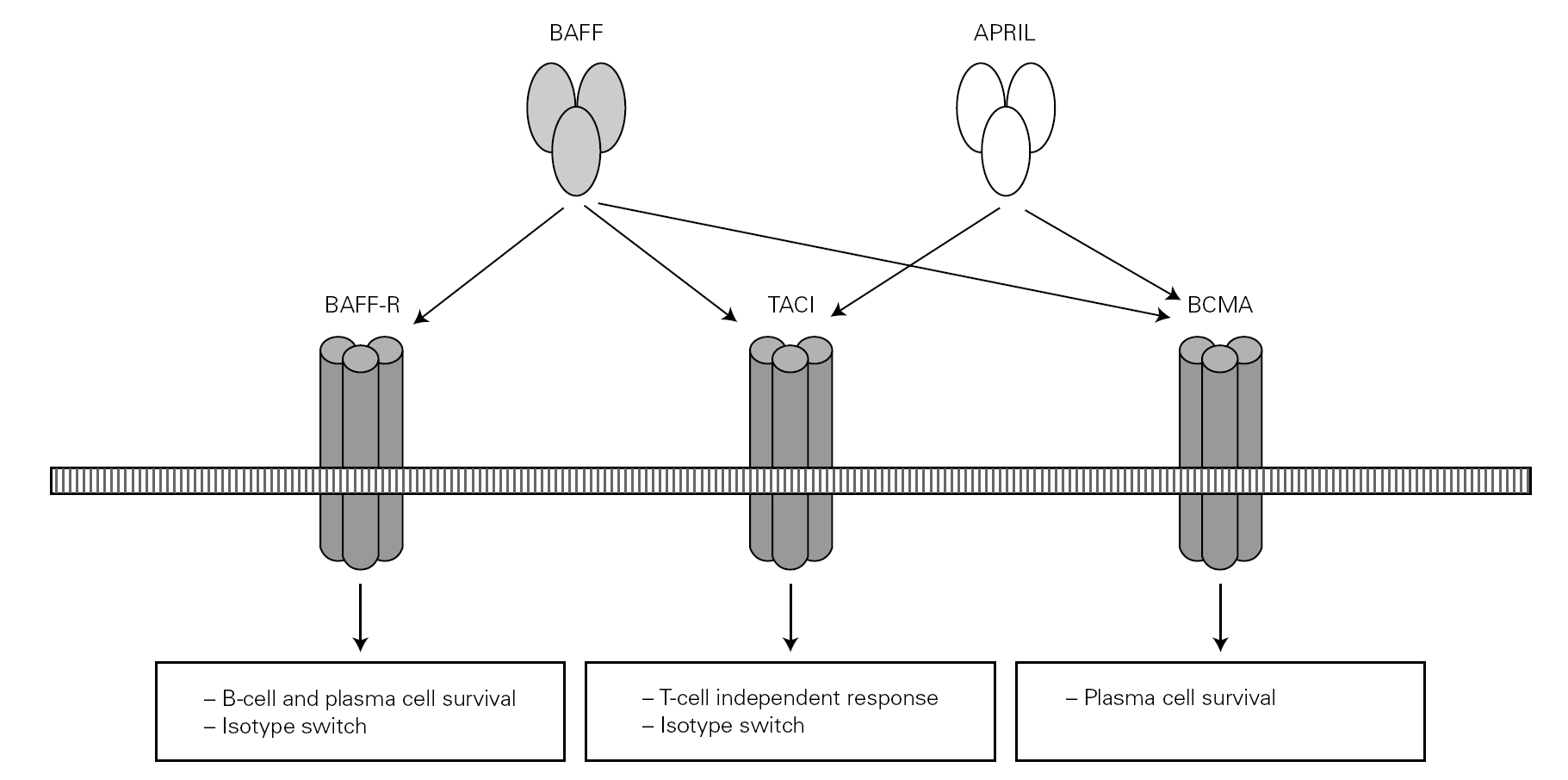
Figure 2.--BAFF (also BLyS or zTNF4) and APRIL factor and the receptors on the cell surface. The proteoglycan receptor is not shown. The functions derived from cell activation are different according to the engage receptor involved. BAFF: B cell activating factor of the TNF family; APRIL: a proliferation inducing ligand; BAFF-R: BAFF receptor; TACI: transmembrane activator and calcium-modulator and cyclophilin ligand interactor; BCMA: B cell maturation antigen.
BAFF factor
The BAFF molecule (also known as BLyS or zTNF4) is encoded for by a 6-exon gene located in 13q34 74. It is synthesized by antigen-presenting cells (APCs), dendritic cells and monocytic cells, and also by neutrophils. IL-10, IFN-γ and IFN-α are potent stimulators of BAFF expression 75. Its principal function is to prolong B lymphocyte life, thus increasing the available B cell population. To this effect, BAFF factor acts upon the cell cycle molecules with participation in cancer processes, such as Bcl-2, Pim or p53. Curiously, the BAFF and p53 genes are very close to each other (a mere 200 kb).
The increase in cell survival is only exerted upon certain partially mature B lymphocytes that have emerged from the bone marrow and are located in the spleen and lymphoid follicles. The factor possibly also acts upon mature plasmocytes, though action upon the particular population of peritoneal B1 lymphocytes has been discarded 76. In sum, BAFF supplies the body with a numerous B cell population. The selectivity of this action, targeted to partially mature subpopulations, is fundamental since an increased survival of marrow B cells (more immature and difficult to control) would increase the risk of autoimmune phenomena and tumors 76.
BAFF also activates non-immune cells, and an excess in its synthesis induces autoimmunity in transgenic mice 77. High serum BAFF levels have been reported in humans with autoimmune or inflammatory diseases such as systemic lupus, rheumatoid arthritis, myasthenia gravis, and particularly Sjögren's disease 78,79. This finding opens up new pathogenic and therapeutic perspectives for these illnesses.
APRIL factor
Although APRIL factor and BAFF factor have 50 % protein homology, and moreover share receptors, their functions are not the same. APRIL factor does not intervene in B cell survival 80, though an influence upon T lymphocytes is not ruled out. Its principal function is oncogenic, not immune with expression in different tumor lines, particularly glioblastoma 81. In addition, it has been speculated that blockade of APRIL factor could be of therapeutic utility 76.
Receptors
The BAFF and APRIL factors bind to three different receptors (BR3, TACI and BCMA) belonging to the TNF receptor superfamily (TNFRSF), and which are found on the surface of B lymphocytes though TACI is also weakly expressed by other cells, such as activated T lymphocytes 76. Binding to these receptors induces different actions related to the maturation and survival of B lymphocytes 82.
The TACI receptor is a molecule encoded for by a 5-exon gene located in 17p11.2, containing two cysteine-rich domains where the TNF-type molecules bind, and moreover facilitating the interbonding of several TACI molecules their prior trimerization or oligomerization being necessary in order to behave as a receptor and activate the cell. The intracytoplasmic portion of the TACI molecule activates the nuclear factor of the activated T cells (NF-AT) following a long metabolic route involving the participation of JNK (c-Jun NH2-terminal kinase) and nuclear factor NFkB. The B subpopulation located in the marginal zone and the CD27 + memory cells are those that express TACI most intensely 83.
Deficiencies in mice
The activator molecules partially share their receptors, which explains the fact that the consequences of the elimination of a molecule or receptor in a transgenic mouse are different.
In mice lacking BAFF factor, a serious block of B lymphocyte maturation is observed, and these cells moreover have a much shortened half-life. Antibody synthesis is strongly deficient for both the thymus-dependent and thymus-independent systems 84. The lack of BAFF receptor (BR3) induces a similar though less intense phenotype, with a normal production of IgA antibodies thus suggesting that synthesis of the latter is mainly dependent upon the TACI receptor, which compensates the defect.
Mice lacking APRIL present B cells with normal counts and survival. However, the switch to IgA is seen to fail, and there is no IgA antibody response following oral challenge 80. In contrast, a lack of BCMA does not appear to alter either antibody synthesis or the switch to IgA.
The transgenic mice without TACI, some experiments have revealed the presence of adenopathies and splenomegalia, with a notorious increase in B lymphocytes 85, since it seems that the TACI molecule normally emits apoptotic signals of relevance for homeostasis of the B cell population. These deficient mice present a deficient thymus-independent humoral response; of particular severity is their inability to produce antibodies against bacterial polysaccharides, and upon aging, over 15 % of the animals develop autoantibodies, lupus, glomerulonephritis and lymphoproliferative alterations 83.
Deficiencies in humans
The function of these molecules in humans remains unclear, and the findings moreover coincide only partially with those obtained in mice being more akin to those recorded in certain monkeys 86. The murine anomalies are more intense than in humans, possibly due to the transgenic model itself, or because humans have acquired alternative functional routes. The TACI molecule belongs to the TNF receptor superfamily (TNFRSF), and in humans several inflammatory or immune diseases are known, attributable to alterations in this group of molecules. Thus, TNFRSF1A mutations cause TNF receptor associated periodic fever syndrome (TRAPS), which exhibits a dominant autosomal hereditary pattern 87-89. Mutations affecting TNFRSF5, commonly referred to as CD40, are responsible for the type 3 (recessive autosomal) presentation of hyper-IgM syndrome 90-92. Mutations affecting TNFRSF6, also called FAS, induce autoimmune lymphoproliferative syndrome (ALPS) a special type of immune deficiency with lymphoproliferation 93,94.
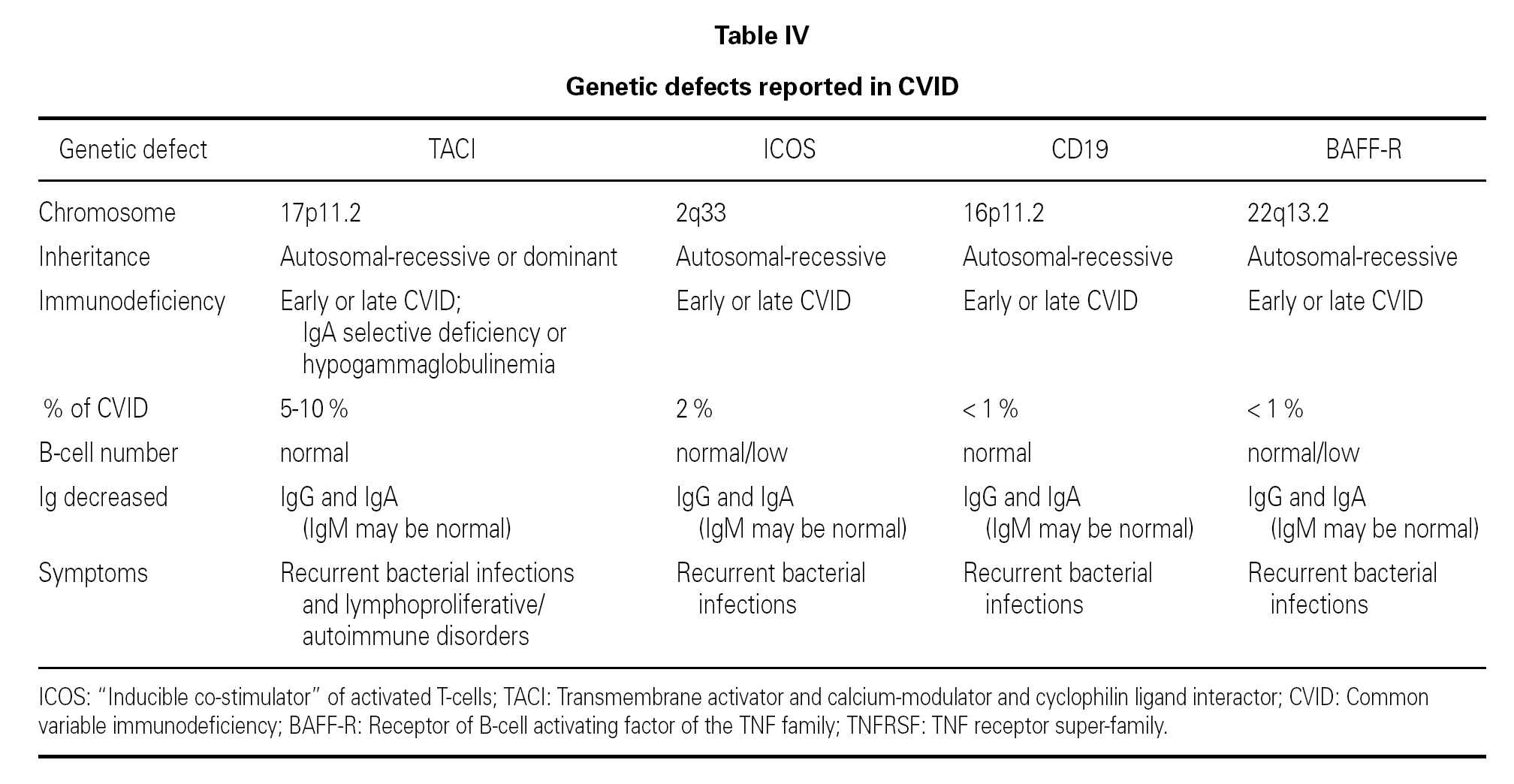
CVID with TACI defect
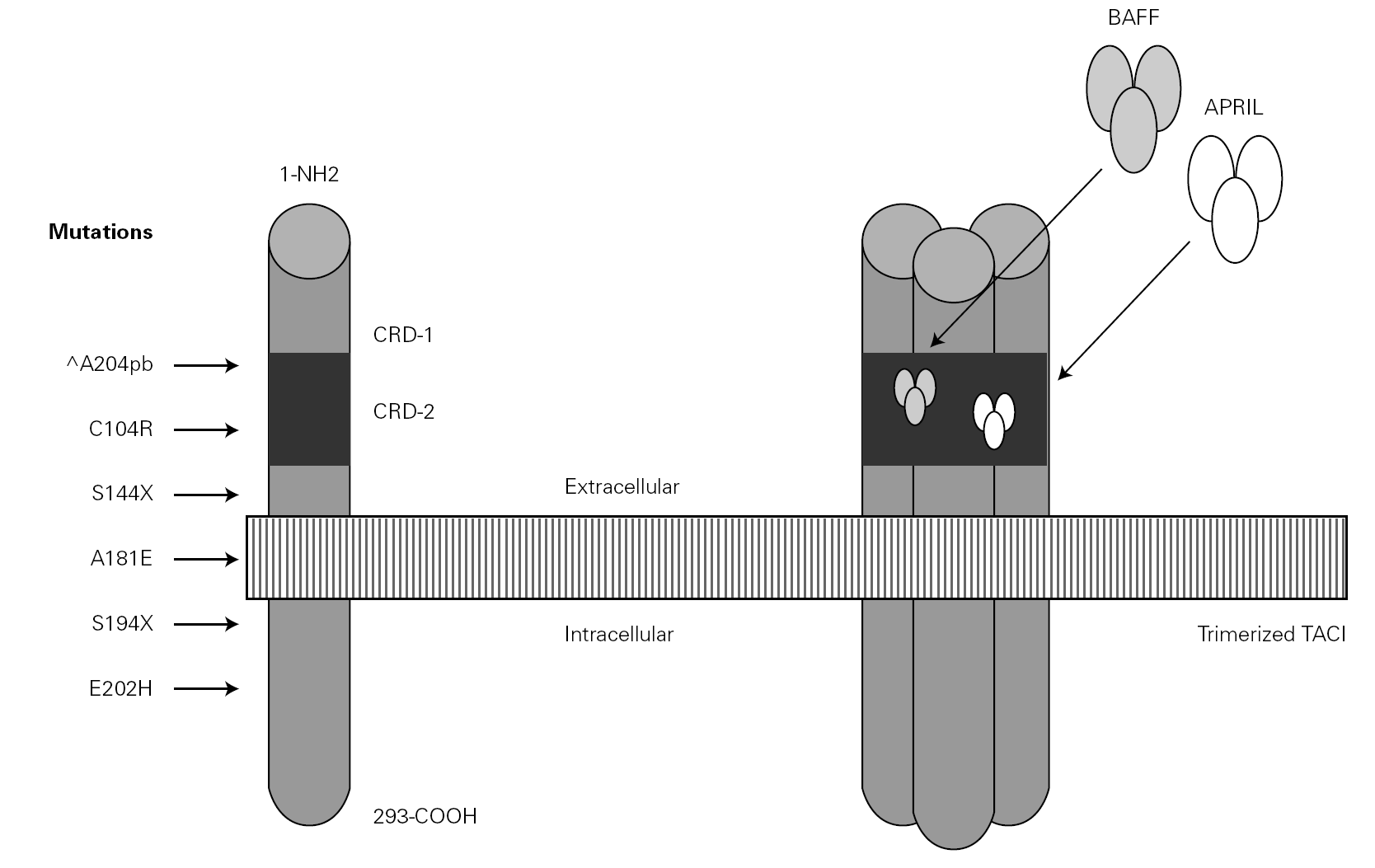
In the year 2005, a group in Europe and another in Boston, respectively directed by Grimbacher 74 and Geha 72, simultaneously published several cases of CVID and of IgA deficiency with mutations of the TNFRSF13B gene, which encodes for the TACI molecule. The findings of both groups were similar, and the mutations identified coincided (S144X, C104R, A181E, S194X and R202H) and appeared in both sporadic forms and in familial presentations though never in normal controls 86. The B lymphocytes of the ill patients expressed TACI, but were unable to synthesize either IgG or IgA in response to the corresponding ligand (APRIL) (72) (fig. 3).
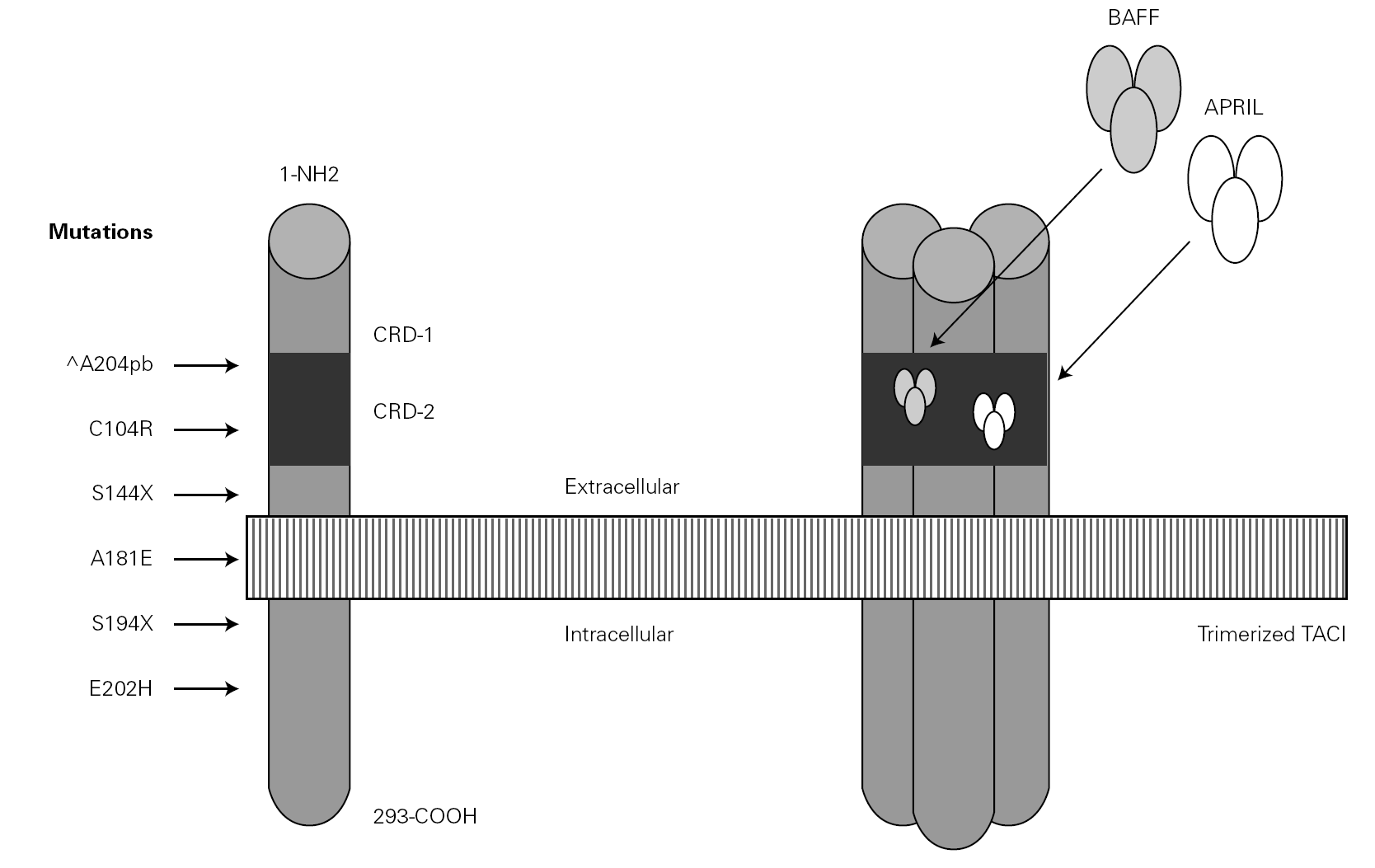
Figure 3.--Structure of the TACI receptor. The two ligands are bound by the cysteine-rich domain-2 (CRD-2). Molecular oligomerization occurs when the receptor is activated. Six mutations have been described in the TACI gene, located in 17p11.2; two of them affect CRD-2.
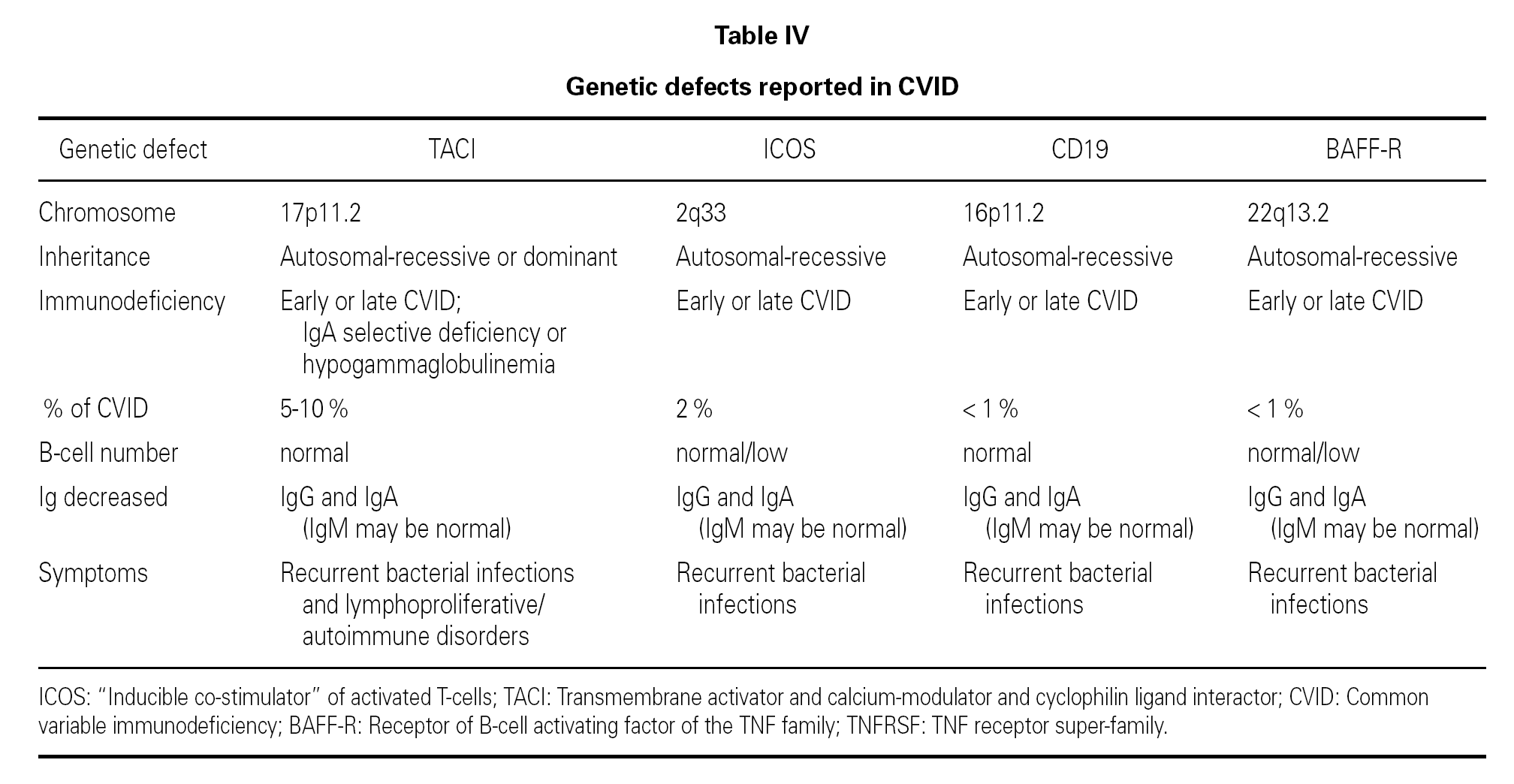
An observation of note is the fact that there were cases in homo- and heterozygosis, and although some of the former presentations exhibited a more serious phenotype, this was not always the case 83. The S144X mutation was associated to the cases that were more serious and more similar to the findings in knock-out transgenic mice, though it also produced asymptomatic hypogammaglobulinemia and never an increase in the B cell population, as in mice 74 (table IV).
In several families, the same mutation caused selective IgA deficiency in some individuals and CVID in the rest 75. The variable penetrance of the deficiencies means that in addition to the actual mutation, other environmental or genetic factors influence the immune and clinical alterations 72,95, and that the activation system in which the TACI molecule participates is highly redundant in humans 74. The majority of cases of CVID with TACI defect reported to date correspond to adults in the 30-70 years age range, with a similar sex distribution. Infectivity was little or slightly increased, and very limited to encapsulated bacteria. The most constant defect was a selective absence of response to polysaccharide vaccination (Pneumovax-23) 72. A little over 30 % showed generally mild autoimmune alterations, or lymphoproliferative processes, usually limited to splenomegalia or tonsillar hypertrophy, and which were only a little more frequent than in the normal population of the same age 83. Such moderation was unexpected, considering the intensity of the findings in transgenic mice.
It has been proposed that cases of CVID with TACI defect should be regarded as an entity independent of the rest of CVID presentations 72, because in all these patients the IgM values were found to be normal this situation not being common in other cases of CVID 41.
Other mutations in CVID
ICOS (inducible costimulatory receptor)
This is a T cell costimulatory factor that facilitates intense IL-10 production and also participates in the synthesis of IL-4, IL-5 and IL-6. Both mice and humans with mutations of the ICOS gene show humoral immune failure compatible with CVID, with anomalous germinal centers 73,96,97. However, the frequency of the ICOS mutation in CVID patients is very low (a little over 1 %) 97,98.
CD19
CD19 regulates the development, activation and proliferation of B lymphocytes 99. Although to date no defects in the so-called co-receptor molecules (CD19, CD21, CD81 and CD225) had been detected in immune deficient patients 100, a recent report describes a homozygous mutation of the CD19 gene in four families with CVID presenting hypogammaglobulinemia and diminished memory B cell and CD5 + B cell counts 73,101. In future, defects of other molecules of this type may appear.
BAFF
On confirming the importance of the BAFF molecule (BLyS) in the development and maturation of B lymphocytes, its encoding gene was considered a candidate for CVID, and has recently been investigated. Losi et al 102 sought mutations in the 6 exons of the gene, though without success. Although mutations of the BAFF gene in CVID have not been ruled out, their frequency would be very low 102. Moreover, and in contrast to the TACI gene, the BAFF gene is highly preserved and shows scant variability 98. To date, BAFF mutations have only been found in patients with systemic lupus erythematosus or rheumatoid arthritis, though with a frequency insufficient to associate them with an increase in susceptibility 103.
BR3 or BAFF-R
The BAFF receptor is important for the development and survival of B lymphocytes. A mutation of the BAFF-R gene was detected in a patient with CVID, though it also appeared in a healthy relative thus raising doubts as to its potential role 73,98.
Correspondence:
Prof. Alfredo Blanco-Quirós
Departamento de Pediatría
Facultad de Medicina
Ramón y Cajal, 5.
47005 Valladolid. Spain
Fax: + 34-983-183812
E-mail: ablanco@ped.uva.es
*Coordinator of Section: Dra. M. Fernández Benítez.
