



INTRODUCCION
Hay evidencia del incremento de la prevalencia del asma en la infancia (1-9). La alergia a hongos y en concreto la sensibilización a Alternaria se considera una de las más importantes causas de rinitis alérgica y asma mediada por IgE, en Europa y EE.UU. Parece ser la causa más común de sensibilización alérgica en niños, inmediatamente después de los ácaros del polvo y del polen de gramíneas (1, 8-15).
La Alternaria se puede encontrar de forma casi constante en el aire, presentando dos máximos en la mayoría de las provincias andaluzas, uno a finales de primavera y otro en otoño (16-19). De los varios alergenos de Alternaria identificados el principal es Alt a 1, que ha sido purificado por varios grupos de investigadores (20-24).
La eficacia de la inmunoterapia subcutánea con vacunas estandarizadas para tratar la alergia a hongos ha sido demostrada en los estudios llevados a cabo por Malling en adultos, y por Dreborg en niños frente a Cladosporium, así como los de Hørst frente a Alternaria (15, 25-27).
Sin embargo, la seguridad de la inmunoterapia subcutánea se ha cuestionado en los últimos años (28-38), por lo que, lograr la misma eficacia, mejorando la comodidad de administración (importantísimo en niños), reduciendo el coste y disminuyendo las reacciones adversas, tanto locales como sistémicas, hace que cobre gran importancia la inmunoterapia local (25, 39).
En el Artículo de Opinión de la OMS sobre la Inmunoterapia Local, de 1998, se acepta la inmunoterapia por vía nasal y sublingual como posibles opciones terapéuticas. En cuanto a la inmunoterapia oral, la OMS opina que no hay documentación suficiente que demuestre su eficacia, por lo que son necesarias ulteriores investigaciones para su validación (39).
Siguiendo las recomendaciones de la OMS, ante la escasez de trabajos realizados en niños asmáticos, y el alto porcentaje de ellos que presentan sensibilización a hongos, como Cladosporium y Alternaria, y la alta incidencia de reacciones adversas a los extractos de Alternaria en su administración subcutánea, se planteó la realización de un ensayo clínico para valorar las repercusiones clínico biológicas de una vacuna oral de Alternaria.
El objetivo es valorar la eficacia clínica de la inmunoterapia de forma directa, mediante la puntuación de síntomas y medicación, recoger la incidencia de reacciones adversas y analizar la repercusión a nivel bronquial, cutáneo y humoral.
MATERIAL Y MÉTODOS
Pacientes
Durante un año se llevó a cabo la selección de los pacientes entre los que acudían a la Unidad de Alergia e Inmunología del Hospital Universitario Reina Sofía de Córdoba.
Cuarenta y cuatro pacientes con edades comprendidas entre 7 y 17 años fueron incluidos en el estudio en base a los siguientes criterios: 1) Historia clínica compatible con asma y/o rinoconjuntivitis inducida por hongos. 2) Sensibilización específica a Alternaria alternata, sola o asociada a pólenes y/o epitelios, demostrada mediante IgE específica y prick test positivo. 3) Test de broncoprovocación con extracto de Alternaria, positivo.
Como criterios de exclusión se contemplaron: 1) Padecer enfermedades inmunológicas sistémicas. 2) Dermatitis atópica grave. 3) Asma grave que requiriera medicación diaria. 4) Tratamiento crónico con corticoides. 5) Haber recibido inmunoterapia con extractos de hongos en los 2 años previos al estudio.
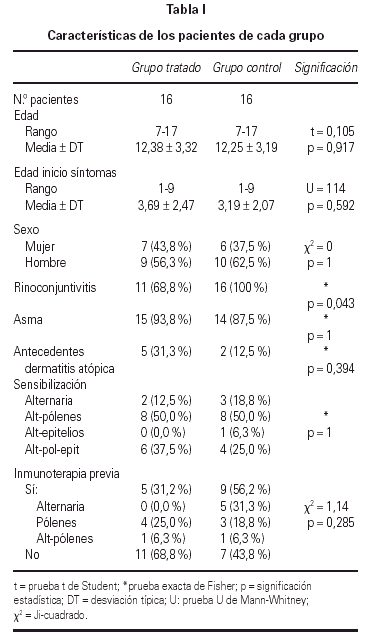
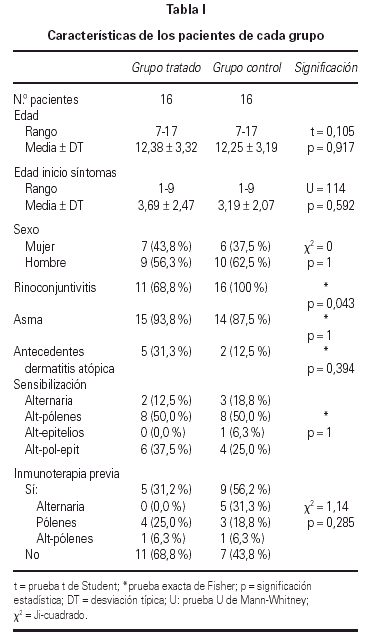
De los 44 pacientes seleccionados, completaron el estudio 39, los 5 restantes lo abandonaron por los siguientes motivos: 3 por requerir medicación continua, 1 por falta de colaboración, 1 por pasar a ser controlado por el servicio de Neumología. Todos los pacientes seleccionados o sus familiares fueron verbalmente informados, obteniéndose de los incluidos en el estudio su conformidad. Las características de los pacientes de cada grupo se detallan en la tabla I.
Vacuna alergénica
La vacuna utilizada "Alergo Merck Oral Alternaria" procede de Merck Farma y Química, S.A., Barcelona (España), preparadas a partir del extracto alergénico producido por Allergopharma, Reinbek (Alemania), la cual fabrica y estandariza los extractos siguiendo las normas del Consejo Nórdico de Medicina.
La actividad alergénica del extracto fue determinada mediante RAST inhibición usando discos de papel (40). La cuantificación del alergeno principal (Alt a 1) se llevó a cabo mediante la técnica de fijación en dos lugares, utilizando anticuerpos monoclonales específicos por radioinmunoensayo, siendo el contenido medio 34,2 ng Alt a 1/μg de proteína (41).
Pauta de inmunoterapia
Se indicó tratamiento con inmunoterapia a 22 de los 44 pacientes del estudio durante 12 meses. El tratamiento inicial se efectuó con 3 frascos de concentración creciente hasta alcanzar la dosis máxima, que correspondía a 28 gotas del "frasco 3", esta dosis se administró 3 veces por semana como mantenimiento que correspondía a 29.848 PNU/mes. La ingestión de la vacuna se realizó en el domicilio del paciente por la mañana en ayunas, en medio vaso de agua.
Después de cualquier interrupción se calculó la dosis de reinicio, dependiendo de los días que había estado sin tomarla. Si la interrupción se había producido estando en dosis máxima se incrementarían las gotas de 2 en 2, en el resto de los supuestos se realizaría el ascenso de 1 en 1 gota:
1. De 1-20 días sin vacuna →1 gota menos por día sin tomarla.
2. De 21-30 días sin vacuna →1,5 gotas menos por día sin tomarla.
5. De 31-40 días sin vacuna →2 gotas menos por día sin tomarla.
4. Más de 40 días sin vacuna → comenzar de nuevo.
Puntuación de síntomas y medicación
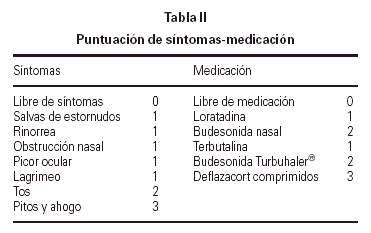
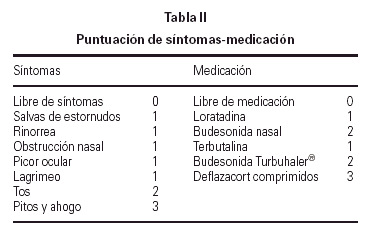
En la primera cita tras la selección, se instruyó a los pacientes o a sus padres sobre la utilización de una hoja estandarizada para la recogida de síntomas y de la medicación que debían tomar para controlarlos, realizando las anotaciones de forma diaria. Los síntomas se agrupaban como síntomas naso-oculares (salvas de estornudos, rinorrea, obstrucción nasal, picor nasal, picor ocular y lagrimeo) y síntomas bronquiales (tos, pitos y ahogo en el pecho).
La medicación utilizada durante el estudio para los síntomas naso-oculares fue loratadina 5-10 mg/24 h durante 3-4 días. Si a pesar de tomarla persistían los síntomas nasales, debían añadir budesonida en spray nasal 100-200 μg/24 h, y mantener ambos medicamentos al menos una semana o hasta la completa remisión de los síntomas.
La utilización de la medicación para el control de los síntomas bronquiales, dependía de la zona de pico de flujo en que se encontrase (verde: > 80 % de su PEF, amarilla: 60-80 % de su PEF y roja: < 60 % de su PEF) que previamente se había calculado para cada paciente a partir de su mejor valor personal. La medición del pico de flujo la llevaba a cabo al levantarse y al acostarse, anotando en una gráfica la mejor de las tres realizadas.
La medicación utilizada al entrar en zona amarilla fue terbutalina sulfato 0,5-1 mg/6-8 h en sistema Turbuhaler®, durante 3-4 días. Si no volvía a zona verde, debía añadir budesonida 200-400 μg/12 h en sistema Turbuhaler® como mínimo durante una semana o hasta llevar 5-6 días en zona verde.
La medicación utilizada al entrar en zona roja era también terbutalina sulfato, pero al doble de dosis de su pauta habitual, además de añadir deflazacort ¼ mg/kg durante 7-10 días. Si su PF no subía a zona amarilla a los 30 min, debía contactar con la Unidad de Alergia o acudir a urgencias para mejor valoración y tratamiento.
En las revisiones efectuadas cada 3 meses en la Unidad de Alergia, se procedía al recuento del número de días que habían presentado síntomas y a la asignación de una puntuación tanto para los síntomas como para la medicación (tabla II), calculándose la puntuación media diaria durante 3 meses al inicio y final del estudio, así como la puntuación media diaria por trimestres naturales. En el grupo con inmunoterapia, los 3 meses iniciales correspondieron al período con pauta de ascenso, para poder mantener la dosis máxima durante 12 meses.
Tests cutáneos
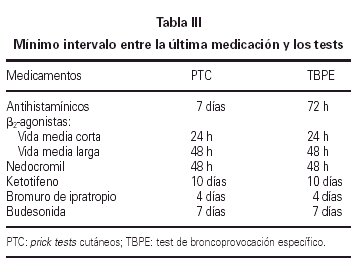
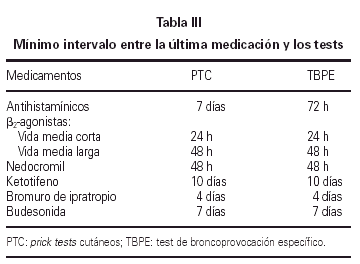
El test cutáneo a punto final se realizó por duplicado siguiendo el método modificado de Pepys y de la EAACI (42). La medicación que pudiera alterar la respuesta cutánea fue suspendida con la suficiente antelación (tabla III). En el día del test se prepararon 4 diluciones a partir del extracto de Alternaria alternata (50.000 SBU/ml) que contiene 3,7 μg/ml de Alt a 1, a una concentración de 50, 500, 5.000, 50.000 SBU/ml. Como control positivo se utilizó la histamina a 10 mg/ml, como control negativo solución salina normal.
La lectura se hizo a los 15 min. Se marcó el perímetro de la pápula formada con un rotulador fino y se pasó a papel adhesivo transparente. Con una regleta milimetrada se midió el diámetro mayor y el perpendicular a él, calculándose el área de la misma. El cambio en la sensibilidad cutánea de cada paciente durante el año de estudio, se calculó para la 4.ª dilución (50.000 SBU/ml), mediante la diferencia de porcentaje entre el tiempo inicial (T0) y tras 12 meses de tratamiento (T1), comparándola con la pápula de histamina considerada como el 100 %.
TBPE
El test de broncoprovocación específica fue realizado siguiendo las indicaciones de Aas (43) y de la SEAIC (44). Se tuvieron en cuenta los posibles factores que afectan a la respuesta bronquial, suspendiendo la medicación con el intervalo que se especifica en la tabla III.
Se utilizó un nebulizador BIRD Mark 7 con liberación de partículas entre 0,5-5 mm de diámetro y un flujo medio de 0,9 l/s. El parámetro elegido para valorar los cambios del test fue el flujo espiratorio en el primer segundo, registrado mediante un espirómetro CS Schatzmann US 22.
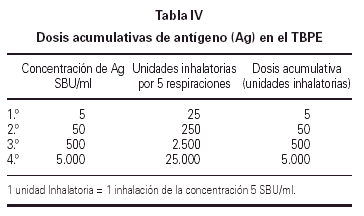
Se emplearon 4 diluciones crecientes de antígeno liofilizado de Alternaria de 5, 50, 500 y 1.000 SBU/ml, preparadas inmediatamente antes de la prueba, comenzando por el diluyente control (solución salina al 0,9 %). Después de la nebulización de cada dilución se realizaba una espirometría a los 10 min, hasta lograr un descenso del FEV1 igual o superior al 20 % del valor obtenido con el diluyente. Tras finalizar el test se administraba el broncodilatador β-agonista de rescate. La respuesta tardía se bloqueó mediante la administración de metilprednisolona i.m. a dosis de 2 mg/kg tras finalizar la fase inmediata del test.
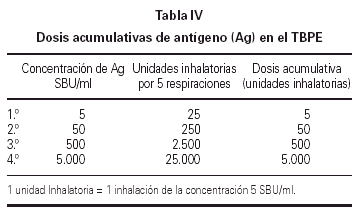
El resultado de la prueba de provocación se expresó mediante la PD20. Este parámetro se suele expresar en forma de Unidades Inhalatorias (UI). En nuestro estudio para simplificar el cálculo de la dosis acumulativa, a 1 UI le hemos dado el valor de 5 SBU/ml (tabla IV).
Función pulmonar
La función pulmonar se controló mediante la medición del pico de flujo (PF) que efectuaba diariamente el paciente, por la mañana y por la noche, con el medidor de pico de flujo (PFM), anotando el mejor valor obtenido de los tres realizados, quedando reflejado en una gráfica que se les proporcionaba y que debían traer a las revisiones trimestrales.
Se calcularon para cada paciente las zonas de PF (verde: > 80 % de su PEF, amarilla: 80-60 % de su PEF, roja: < 60 % de su PEF), a partir de su mejor valor personal. También se calculó el PF medio durante los 3 meses al inicio y final del estudio, teniendo en cuenta que para los pacientes con inmunoterapia el primer trimestre tras su incorporación correspondió al período con pauta de ascenso.
Parámetros inmunológicos valorados
En los dos tiempos de estudio T0 y T1 se procedió a la extracción de sangre de los pacientes por punción venosa. Tras su centrifugación, el suero fue almacenado a 20 °C hasta ser analizado por el laboratorio de Inmunología del Hospital Reina Sofía, para la determinación de los parámetros inmunológicos: IgE total, IgE específica e IgG4 específica mediante fluoroinmunoensayo, empleando el sistema UniCAP 100 de Pharmacia & Upjohn Diagnósticos.
Diseño del estudio
Se llevó a cabo un estudio aleatorio controlado, de 12 meses de duración. Se formaron dos grupos de comparación entre los 44 pacientes seleccionados, mediante la asignación aleatoria individual de los participantes al grupo tratado (22 pacientes), o al grupo control (22 pacientes). Debido a las pérdidas por los motivos especificados en el apartado pacientes, los grupos quedaron constituidos por 19 tratados y 20 controles.
Las variables relevantes, distintas a la pertenencia al grupo tratado o control, como la edad y la sensibilidad específica de cada paciente, se distribuyeron de forma semejante en los grupos de comparación, para que no pudieran interferir en los resultados. Cada paciente pasaba revisiones periódicas cada 3 meses a partir de su inclusión en el estudio (3, 6, 9 y 12), que pretendían supervisar la evolución del mismo y recoger los datos referentes al score de síntomas-medicación, pico de flujo y control del tratamiento médico e inmunoterápico.
Además del seguimiento clínico trimestral al inicio del estudio (tiempo 0, T0) y tras 12 meses de tratamiento (tiempo 1, T1), se efectuó una evaluación de la hiperreactividad específica, con la realización de test cutáneos a punto final con Alternaria y test de broncoprovocación específica, y una evaluación humoral. Para el grupo tratado con inmunoterapia se contaron los 12 meses a partir de haber alcanzado la dosis máxima. Para el grupo control, a partir de su inclusión en el estudio.
Análisis estadístico de los datos
Análisis descriptivo con el cálculo de frecuencias absolutas y relativas porcentuales para las variables cualitativas; las variables cuantitativas se expresaron como media ± desviación típica. En las representaciones gráficas se utilizó la media y su intervalo de confianza al 95 % de seguridad (IC 95 %).
Las comparaciones de porcentajes (variables cualitativas) entre los 2 grupos se realizaron mediante pruebas Ji-cuadrado para tablas de contingencia; para tablas 2 ×2 se utilizaron el estadístico Ji-cuadrado con corrección de Yates, y si alguna frecuencia esperada era menor de 5 se aplicó la prueba exacta de Fisher.
Las puntuaciones de síntomas y medicación se representaron mediante el valor de la mediana y rango intercuartílico y gráficamente mediante un diagrama de caja y bigotes (box-plot). Para detectar si hay diferencias significativas entre el grupo control y el tratado con inmunoterapia se usó la prueba U de Mann-Whitney. Las comparaciones intragrupo (antes-después) se realizaron mediante la prueba de Wilcoxon.
Para comparar las medias aritméticas (variables cuantitativas) de los 2 grupos se empleó la prueba paramétrica t de Student, previa comprobación de la normalidad de las distribuciones mediante el test de Shapiro y Wilks; en caso contrario se utilizó la prueba U de Mann-Whitney. Las comparaciones intragrupo se realizaron con la prueba t de Student para datos apareados. En el caso de que las diferencias de los valores al inicio y al finalizar el estudio no siguieran una distribución normal se empleó la prueba Z de Wilcoxon.
La relación entre las variables cuantitativas se determinó mediante el coeficiente de correlación lineal de Pearson. La relación entre una variable cualitativa ordinal y una cuantitativa se realizó mediante el coeficiente por rangos de Spearman. En todas las pruebas estadísticas se consideraron como significativos los valores de p inferiores a 0,05; los contrastes de hipótesis serán bilaterales. El procesamiento, tratamiento y análisis de los datos se realizó con el programa estadístico SPSS para Windows (versión 8.0).
RESULTADOS
Se realizó el análisis estadístico de los datos en 32 de los 39 pacientes incluidos en el estudio; debido a que sólo 16 de los pertenecientes al grupo tratado consiguieron alcanzar y mantener la dosis máxima, siendo excluidos los restantes así como sus respectivas parejas, quedando 16 pacientes en el grupo tratado y 16 pacientes en el grupo control. No se encontraron diferencias significativas entre ellos antes del tratamiento.
Protocolo de inmunoterapia
Hubo 3 pacientes que no consiguieron alcanzar la dosis máxima, debido a reacciones adversas con el reinicio; el resto la alcanzaron por término medio en 3 meses y la mantuvieron durante 10 a 12 meses, alcanzando unas dosis acumuladas medias de 288.000 PNU, lo que corresponde a 3 o 4 veces la dosis administrada de Alternaria por vía subcutánea.
Tolerancia
En general, el extracto fue bien tolerado. Hubo un total de 8 reacciones adversas (RA) equivalentes a 0,42 RA por 100 dosis administradas. Fueron de carácter leve-moderado y cedieron con el tratamiento médico administrado en domicilio. Tres de ellas se presentaron en la fase de ascenso de la inmunoterapia, consistieron en 1 episodio de broncospasmo y 2 de rinoconjuntivitis-broncospasmo, ocurridas con el reinicio de la vacuna tras interrupción por circunstancias intercurrentes, como infecciones respiratorias. Las 5 restantes, se presentaron en la fase de mantenimiento: 2 episodios de broncospasmo, 1 de rinoconjuntivitis y diarrea en 2 ocasiones.
Hubo 7 abandonos (36,8 %) del total de los pacientes tratados con IT. Los motivos fueron los siguientes: en 5 de ellos se debió a broncospasmo con el reinicio de la vacuna tras una interrupción superior a 2 semanas; en los 2 restantes a broncospasmo tras infección respiratoria aguda. El 15,8 % de los abandonos se dieron en la fase de ascenso de la inmunoterapia, y el 21 % en la fase de mantenimiento.
No se encontró relación significativa entre las reacciones adversas y la dosis acumulada; tampoco con las sensibilizaciones de cada paciente.
Puntuación de síntomas-medicación
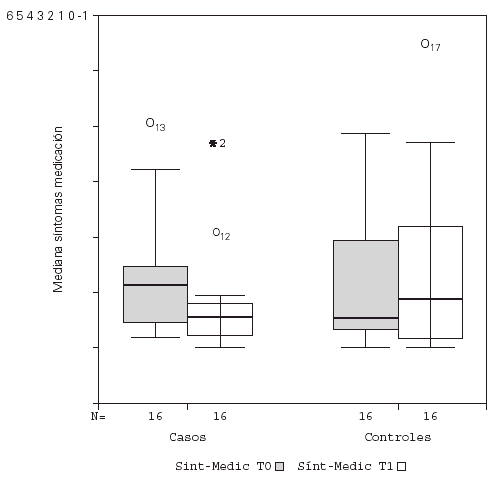
Se detectó un descenso significativo en la puntuación media diaria de "síntomas" y de "síntomas-medicación" al comparar el inicio y final del estudio en los pacientes tratados con inmunoterapia, permaneciendo sin cambio el grupo control, como se observa en la figura 1. No hubo diferencias significativas en la puntuación media diaria de la "medicación", tanto para el grupo tratado como control. Estos resultados se recogen en la tabla V.
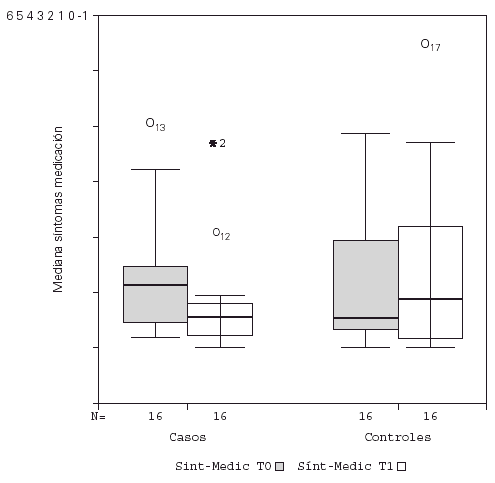
Figura 1.--Descenso significativo de la puntuación media de síntomas-medicación para el grupo tratado con inmunoterapia.

También se analizó la puntuación media diaria de "síntomas-medicación" por trimestres naturales para ambos grupos, ya que se trataba de pacientes polínicos, comprobándose que no hubo diferencias estacionales significativas debidas a otros agentes distintos a Alternaria.
Tests cutáneos
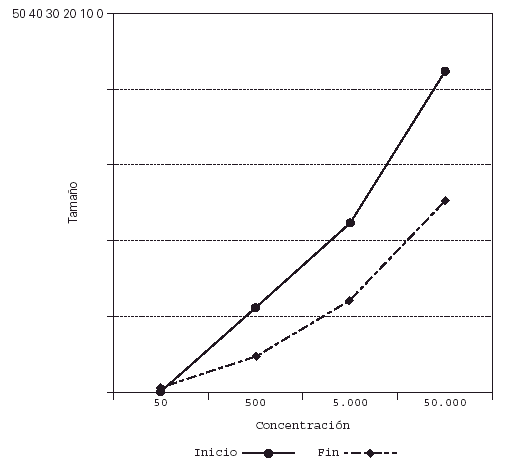
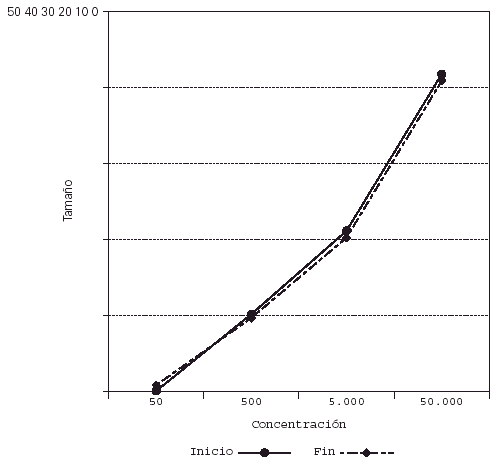
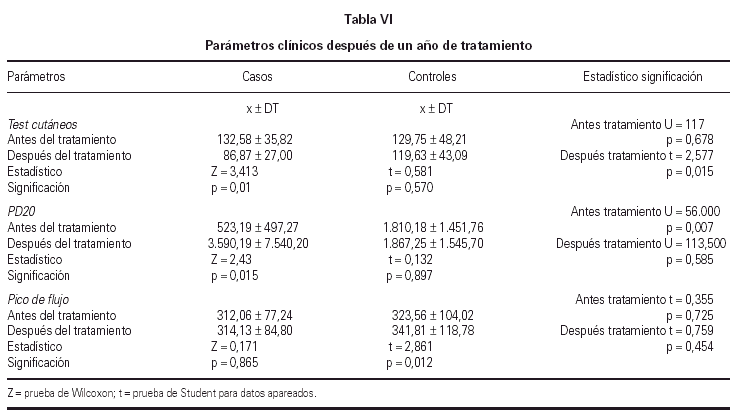
La reactividad de los tests cutáneos disminuyó significativamente en el grupo con tratamiento activo, reduciéndose el porcentaje del tamaño de pápula para la cuarta concentración (tabla VI), no encontrándose diferencias significativas en el grupo control.
Su representación en el sistema de líneas paralelas muestra un desplazamiento a la derecha en el grupo tratado, al haber disminuido el tamaño de pápula para las diferentes concentraciones (fig. 2), no apreciándose desplazamiento en los controles (fig. 3).
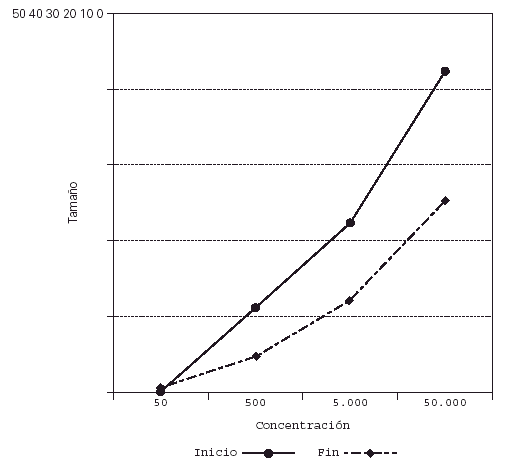
Figura 2.--Casos. Reducción del tamaño de pápula para las diferentes concentraciones al final del estudio.
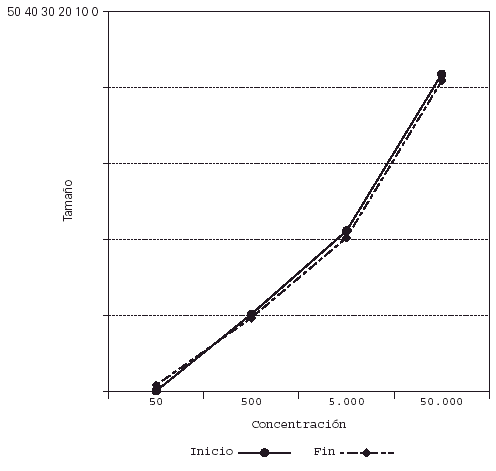
Figura 3.--Controles. Se mantiene el tamaño de pápula para las diferentes concentraciones al final del estudio.
TBPE (PD20)
Se obtuvieron diferencias significativas al comparar el test de broncoprovocación específico expresado mediante la PD20 entre el inicio y final del estudio, con un incremento significativo para los valores de la PD20 en el grupo tratado, manteniéndose en el grupo control (tabla VI).
Función pulmonar
No se apreciaron diferencias significativas entre el pico de flujo medio durante los 3 meses al inicio y final del estudio, tanto para los casos como para los controles. Los datos analizados se recogen en la tabla VI. Tampoco se encontraron diferencias significativas en el número de días que tanto los casos como los controles permanecieron en cada zona.
Inmunidad humoral
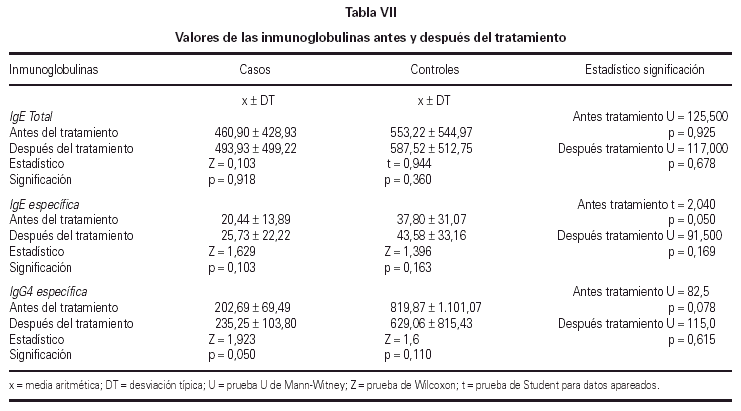
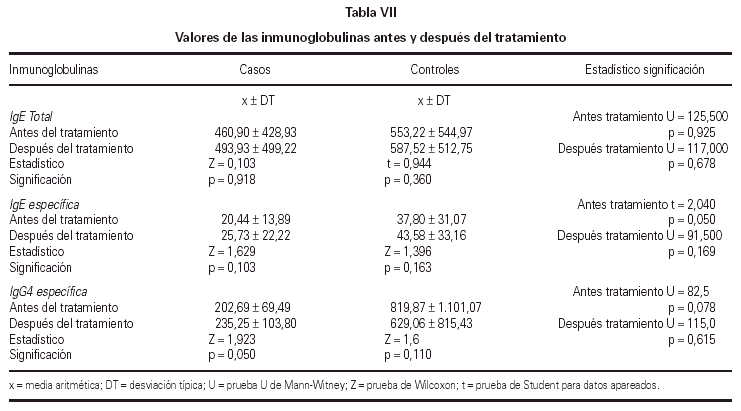
No se encontraron cambios significativos en los valores de IgE total e IgE específica entre el T0 y el T1 de la totalidad de los pacientes. Sin embargo, se observó una elevación significativa de los niveles de IgG4 en el grupo tratado con inmunoterapia oral, al comparar los niveles de la misma antes y después del tratamiento, según se muestra en la tabla VII.
DISCUSION
La evidencia a favor de la inmunoterapia (IT) subcutánea y su efectividad ha dejado de ser materia de debate, siempre que el diagnóstico, administración y seguimiento del paciente sean llevados a cabo correctamente (25). Sin embargo, este método de administración es mal aceptado por la población infantil, además de acompañarse de frecuentes reacciones adversas.
En los últimos años la administración local de alergenos ha despertado un gran interés a todos los niveles, priorizándose la investigación de su eficacia y seguridad. Según Malling (45), el único parámetro que indica eficacia es la reducción de síntomas y/o medicación, disminuyendo significativamente, desde este punto de vista, la morbilidad de las enfermedades alérgicas. Los cambios en los parámetros inmunológicos y en los diversos test pueden ser interesantes a la hora de aportar datos objetivos que aclaren mecanismos de acción.
En este estudio se ha administrado IT específica Alternaria por vía oral (ITO) durante 12 meses, alcanzando dosis acumuladas que superan en 88 % de los casos el triple de las administradas en IT subcutánea.
Al no existir una puntuación estándar aceptada internacionalmente, se ha utilizado una puntuación de síntomas y medicación tanto de forma aislada como global, apreciándose un descenso significativo en la puntuación de síntomas y en la puntuación de síntomas-medicación en el grupo tratado con IT oral Alternaria. Lo que coincide con los hallazgos de otros estudios realizados sobre IT oral con ácaros (46), con polen de abedul (47-49), ambrosía (50) y gramíneas (51), así como en el realizado con IT sublingual con Alternaria (52).
Por otro lado la correlación entre la puntuación de síntomas-medicación al inicio y final del estudio resultó ser significativa, lo que indica que mejora más el paciente que basalmente estaba mejor. Hay que señalar que posiblemente los pacientes polisensibilizados no son los ideales para mostrar el efecto beneficioso de una vacuna, pero constituyen la realidad clínica de las consultas.
En lo referente a seguridad, el porcentaje de reacciones adversas es similar al de otros autores al considerarlo en función del número de dosis administradas, con incremento de síntomas gastrointestinales como dolor abdominal, náuseas y diarrea (47, 50, 53, 54), y episodios de sibilancias y rinoconjuntivitis (48, 55, 56), siendo de carácter leve-moderado y presentándose en su mayoría al reiniciar la vacuna tras interrupciones superiores a 2-3 semanas por circunstancias intercurrentes; tal vez se hubieran beneficiado de una pauta de ascenso aún más lenta que la propuesta.
La mayoría de las reacciones adversas se presentaron en la fase de mantenimiento lo que no coincide con los hallazgos de un estudio multicéntrico llevado a cabo por Almagro et al (69), el cual recogió mayor frecuencia de reacciones adversas en la fase de inicio.
En cuanto a la reactividad cutánea, en el presente estudio ha quedado patente la disminución de la misma en el grupo tratado, valorada mediante la cuarta concentración del test punto final Alternaria. Estos hallazgos coinciden con otros estudios realizados sobre inmunoterapia oral de polen de abedul (48, 49, 58) o inmunoterapia sublingual con ácaros (53, 59), sugiriendo según Hørst (15) la supresión de la liberación del mastocito.
En lo referente al pico de flujo (PF) no se han detectado cambios en los valores recogidos antes y después del tratamiento. Posiblemente sea debido a que se trataba de pacientes polínicos que estuvieron expuestos a diferentes alergenos durante los cambios estacionales, en muchos casos responsables de las interrupciones durante el tratamiento con inmunoterapia.
Sin embargo, se apreció un cambio en la reactividad del órgano diana al tolerar dosis acumuladas de extracto alergénico más elevadas en el TBPE realizado al final del estudio, con PD20 significativamente más altas en el grupo tratado. Este hecho coincide con lo observado por otros autores, tras la administración de inmunoterapia sublingual de ácaros durante 18 y 24 meses, respectivamente (53, 59).
Con respecto a la inmunidad humoral, no hubo diferencias significativas en los niveles de IgE total y específica al comparar el inicio y el final del estudio, al igual que les ocurrió a otros autores que emplearon la IT durante 12 meses (60, 61) o 24 meses (52). Parece ser que al prolongar el tiempo de inmunoterapia de 12 a 24 meses gran número de autores encontraron diferencias significativas en los niveles de inmunoglobulinas (48, 55, 58, 59, 62).
Por otro lado, el papel de la IgG específica, según Malling (63), está implicado directamente en la reducción de la liberación de mediadores por el mastocito. A su vez, su incremento indica activación de las células inmunocompetentes tras la administración de una significativa cantidad de alergeno (45). En nuestro estudio hay una elevación significativa de los niveles de IgG4 en el grupo tratado con IT oral al comparar los valores de la misma antes y después del tratamiento, al igual que lo hallado por Troise (64).
Según lo anteriormente expuesto, concluimos que la inmunoterapia oral con un extracto de Alternaria, ha demostrado eficacia clínica en un grupo de pacientes pediátricos, siendo en general bien tolerada, presentando reacciones adversas exclusivamente de carácter leve-moderado. Ha modificado la reactividad específica cutánea y bronquial, y aunque la dosis acumulada de inmunoterapia recibida no ha sido suficiente para modificar los niveles de IgE total y específica, sí se ha producido una activación de las células inmunocompetentes expresada por la elevación de la IgG4, lo que demuestra la repercusión de la vacuna a nivel humoral.
