


Primary immunodeficiency diseases (PIDs) are a group of heterogeneous inherited disorders, characterised by recurrent infections, autoimmunity and malignancy. Some PIDs such as hyper IgE syndrome (HIES) and Wiskott–Aldrich syndrome (WAS) may be initially presented as atopic dermatitis (AD), especially in its severe form, resulting in diagnostic delay and poor prognosis of patients.
ObjectiveThe aim of this study was to evaluate the frequency of PIDs among patients with severe AD and to determine factors that can help to raise suspicion towards these disorders.
MethodsSeventy-five patients with a well-established diagnosis of severe AD were enrolled in this study. Initial immunological evaluations, including humoral and cellular investigation, were performed in all individuals. Patients underwent further investigations in a case of suspicion of a probable PID.
ResultsAmong all patients with severe AD, five (6.6%) were diagnosed with HIES and one (1.3%) with WAS. Family history of PIDs, family history of death in early infancy, positive history of recurrent infections such as skin and respiratory infections, otitis media and sinusitis were observed significantly higher in patients with a diagnosis of PID.
ConclusionsThe presence of an underlying PID could explain the poor prognosis and refraction to the treatment of some patients with severe AD. Several clinical and laboratory findings can help the physicians to focus towards PIDs which are more serious. Delay in diagnosis of PID cases with skin manifestation of AD without proper management may result in lower quality of life and higher morbidity and mortality rates.
Primary immunodeficiency diseases (PIDs) are inherited disorders that predominantly affected infants and children, which are usually characterised by recurrent infections of different organs as well as autoimmunity and malignancy in some cases.1,2 Skin is one of the organs that can be involved in PIDs and its complications include infections and abscesses (e.g., in severe combined immunodeficiency, chronic mucocutaneous candidiasis, and phagocyte defects), poikiloderma (e.g., in poikiloderma with neutropenia), erythroderma (e.g., in Omenn syndrome), anhydrotic ectodermic dysplasia (e.g., in NEMO deficiency, autosomal-dominant anhydrotic ectodermal dysplasia with immunodeficiency, calcium channel deficiency with ORAI-I deficiency and STIM-1 deficiency), keratinocytes defect with human papilloma virus infections and cancer of the skin (e.g., in epidermodysplasia verruciformis) eczematous dermatitis, petechiae, vasculitis and autoimmune skin disorders (e.g., in predominantly antibody deficiency) which may be the clue for the final diagnosis of a PID.2–5
Atopic dermatitis (AD) is a common eczematous skin disorder which mostly occurs during the infancy and childhood, characterised by intense itchy, erythematous papules, vesicles, and oedema in acute stages and lichenification in chronic stages. It is also associated with increased serum IgE levels.6,7 Patients with the severe form of AD do not effectively respond to conventional treatments of this disorder such as oral antihistamines and topical corticosteroids8; therefore they need systemic immunosuppressive agents and to search for the cause of severity.9,10
Eczematous dermatitis resembling severe AD occurs frequently and is associated with a few types of PIDs, including selective IgA deficiency, common variable immunodeficiency (CVID), Wiskott–Aldrich syndrome (WAS), X-linked agammaglobulinaemia, hyper IgE syndrome (HIES), and Omenn syndrome.4,6,11–13
The presence of a PID in patients with severe AD should be investigated, since severe AD can be the first presentation of these disorders. Timely diagnosis and management of patients with PIDs are of significant importance and delay in diagnosis is associated with higher rates of morbidities and lower quality of life, especially in cases with non-infectious manifestations.14–16
There are no specific data on the prevalence of a PID in subjects presenting with severe AD. Moreover, co-factors that help in clinical suspicion of a PID in severe AD phenotype are not completely identified. We studied patients with severe AD to investigate presence of PIDs in these patients assessing their clinical and laboratory findings.
Patients and methodsPatientsBetween March 2010 and April 2011, all qualified patients who visited the Department of Dermatology at the Razi Hospital and the Department of Immunology at the Children's Medical Center Hospital (Tehran, Iran) with a well-established diagnosis of severe AD were enrolled for immunological evaluations. Both hospitals are affiliated to Tehran University of Medical Sciences. Patients with a known secondary immunodeficiency were excluded from the study.
According to the Hanifin & Rajka criteria,6 the diagnosis of AD is proven with at least three major criteria including: itching, lichenification (flexor and linear surfaces in adolescents and extensor surfaces lichenification in children), chronic or recurrent symptoms, atopy (asthma, allergic rhinitis, atopic dermatitis) past or family history, and three minor criteria including: skin dryness, cheilitis, fissure, etc. AD was defined as severe, when the calculated value of the severity scoring system for AD (SCORAD) became higher than 50 points. The failure to effectively control the clinical condition of the patients by the use of routine therapeutic agents such as oral antihistamines, topical corticosteroids, topical moisturisers and topical calcineurin inhibitors were also considered in these patients.10,17,18 Written informed consents were acquired from all patients or their legal guardian(s) and the process of this study was approved by the Ethics Committee of Tehran University of Medical Sciences.
Immunological evaluationsIn the first visit, a three-page questionnaire, including demographic information and complete medical history was filled for every patient with severe AD. A full clinical examination was performed and laboratory tests, including complete blood cell count (CBC), serum immunoglobulin level (IgA, IgG, IgM and IgE) and lymphocyte CD markers measurement, were done as indicated. Further immunological tests were performed if needed in some cases. The diagnosis of PIDs was made using the standard criteria of the Expert Committee of International Union of Immunological Societies (IUIS) on Primary Immunodeficiencies.2
Statistical analysisThe data were analysed using the SPSS program version 16.0. For the evaluation of immunological data and CD markers, we used results of initial test at the time of diagnosis. Comparisons between groups were performed using Student's t-test for continuous data; Mann–Whitney U-test was used when the distribution was not normal for the selected variable. Chi-squared and Fisher's exact tests were performed between groups to compare clinical presentations as indicated. P-values<0.05 were considered as statistically significant.
ResultsPatients’ characteristicsSeventy-seven individuals with a well-established diagnosis of severe AD completed all stages of this study, but two patients were excluded from the study due to the presence of secondary immunodeficiency (cystic fibrosis and human immunodeficiency virus infections). Finally, a total number of 75 severe AD patients (44 male and 31 female) were investigated for possible underlying PIDs.
The median age of the patients at the time of the study was two years, ranging from four months to 30 years. Seventy-one patients (94.7%) were under 16 years of age and only four patients (5.3%) older than 16 years. The median age of the patients at the onset of AD was 0.3 year. Parental consanguinity was documented in 36 patients (48%) of studied cases. Seventy-two out of 75 patients (96%) had complete vaccination history until the time of study, while the vaccination was not completed in the remaining three patients (4%) due to the presence of an active infection at the time of the scheduled vaccination. Four patients (5.3%) had a positive family history of death with an unknown cause at the childhood.
Twelve out of 75 patients (16%) had a positive history of recurrent infections in different organs, including respiratory system in seven (9.3%), skin in six (8%), ear in eight (10.6%) and sinus in three patients (4%); eight patients (10.6%) had this history in more than one system.
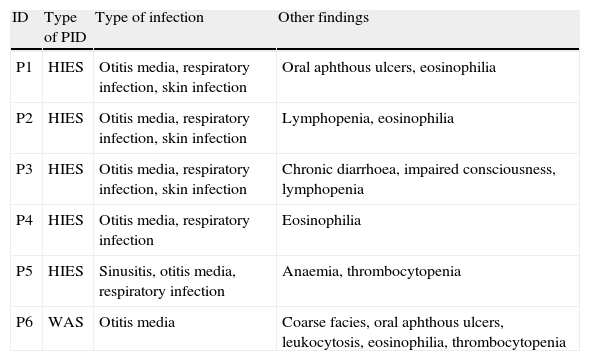
Immunological evaluationThe mean (SD) white blood cell (WBC) count of the patients was 10,696.2 (3988.0)cell/mL. The mean (SD) serum levels of IgG, IgM, IgA and IgE were 670.67 (268.5)mg/dL, 87.52 (34.77)mg/dL, 94.56 (108.6)mg/dL and 393.11 (769.87)IU/mL, respectively. Twenty-nine patients needed further immunological tests, which resulted in definitive diagnosis of PID in six out of 75 patients (8%). HIES was diagnosed in five patients (6.6%), while WAS was the diagnosis of only one patient (1.3%). Characteristics features of six AD patients with the diagnosis of PIDs are shown in Table 1.
Recurrent infections in PID patients with the presentation of severe AD.
| ID | Type of PID | Type of infection | Other findings |
| P1 | HIES | Otitis media, respiratory infection, skin infection | Oral aphthous ulcers, eosinophilia |
| P2 | HIES | Otitis media, respiratory infection, skin infection | Lymphopenia, eosinophilia |
| P3 | HIES | Otitis media, respiratory infection, skin infection | Chronic diarrhoea, impaired consciousness, lymphopenia |
| P4 | HIES | Otitis media, respiratory infection | Eosinophilia |
| P5 | HIES | Sinusitis, otitis media, respiratory infection | Anaemia, thrombocytopenia |
| P6 | WAS | Otitis media | Coarse facies, oral aphthous ulcers, leukocytosis, eosinophilia, thrombocytopenia |
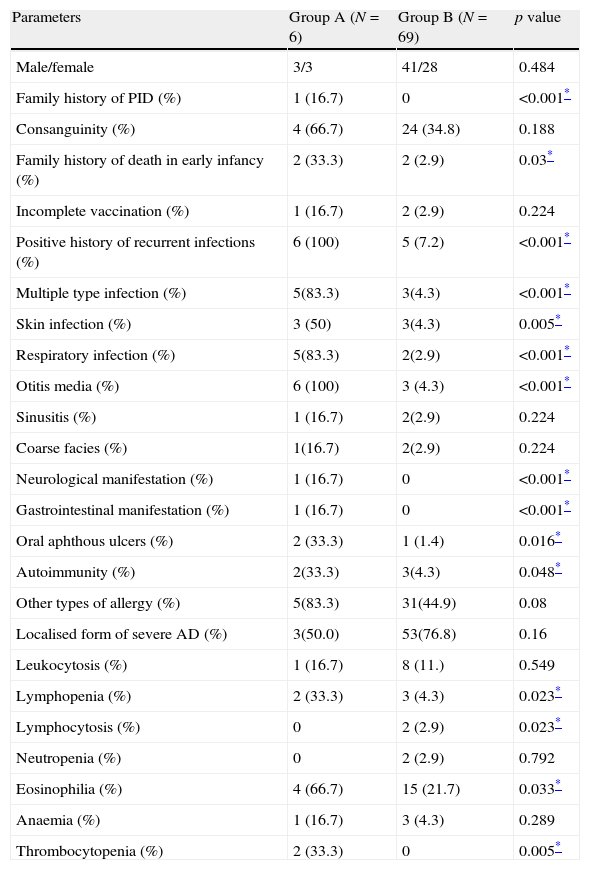
Various clinical and laboratory data were compared between AD patients with a confirmed diagnosis of PID (Group A) and the rest of the AD patients (Group B) in Tables 2 and 3. The family history of PID was positive in one patient (P4, HIES in sister) in Group A (16.7%), while this finding was not observed in Group B (p<0.001). Positive family history of death in early infancy was observed in two out of six patients (33.3%) in Group A, significantly higher than two out of 69 patients (2.9%) in Group B (p=0.03). Positive history of recurrent infections was observed in all patients in Group A, while it was observed in only five out of 69 patients (7.2%) in Group B (p<0.001).
Comparison of various qualities between severe AD patients with an underlying PID (Group A) and those without PID (Group B).
| Parameters | Group A (N=6) | Group B (N=69) | p value |
| Male/female | 3/3 | 41/28 | 0.484 |
| Family history of PID (%) | 1 (16.7) | 0 | <0.001* |
| Consanguinity (%) | 4 (66.7) | 24 (34.8) | 0.188 |
| Family history of death in early infancy (%) | 2 (33.3) | 2 (2.9) | 0.03* |
| Incomplete vaccination (%) | 1 (16.7) | 2 (2.9) | 0.224 |
| Positive history of recurrent infections (%) | 6 (100) | 5 (7.2) | <0.001* |
| Multiple type infection (%) | 5(83.3) | 3(4.3) | <0.001* |
| Skin infection (%) | 3 (50) | 3(4.3) | 0.005* |
| Respiratory infection (%) | 5(83.3) | 2(2.9) | <0.001* |
| Otitis media (%) | 6 (100) | 3 (4.3) | <0.001* |
| Sinusitis (%) | 1 (16.7) | 2(2.9) | 0.224 |
| Coarse facies (%) | 1(16.7) | 2(2.9) | 0.224 |
| Neurological manifestation (%) | 1 (16.7) | 0 | <0.001* |
| Gastrointestinal manifestation (%) | 1 (16.7) | 0 | <0.001* |
| Oral aphthous ulcers (%) | 2 (33.3) | 1 (1.4) | 0.016* |
| Autoimmunity (%) | 2(33.3) | 3(4.3) | 0.048* |
| Other types of allergy (%) | 5(83.3) | 31(44.9) | 0.08 |
| Localised form of severe AD (%) | 3(50.0) | 53(76.8) | 0.16 |
| Leukocytosis (%) | 1 (16.7) | 8 (11.) | 0.549 |
| Lymphopenia (%) | 2 (33.3) | 3 (4.3) | 0.023* |
| Lymphocytosis (%) | 0 | 2 (2.9) | 0.023* |
| Neutropenia (%) | 0 | 2 (2.9) | 0.792 |
| Eosinophilia (%) | 4 (66.7) | 15 (21.7) | 0.033* |
| Anaemia (%) | 1 (16.7) | 3 (4.3) | 0.289 |
| Thrombocytopenia (%) | 2 (33.3) | 0 | 0.005* |
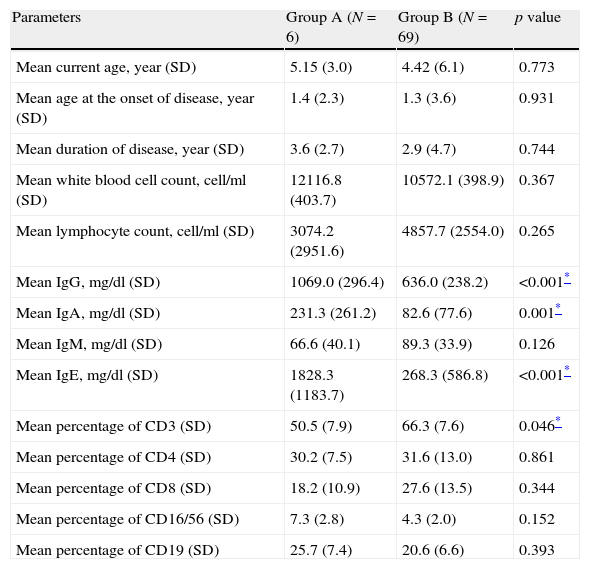
Comparison between various quantitative measurements between severe AD patients with an underlying PID (Group A) and those without PID (Group B).
| Parameters | Group A (N=6) | Group B (N=69) | p value |
| Mean current age, year (SD) | 5.15 (3.0) | 4.42 (6.1) | 0.773 |
| Mean age at the onset of disease, year (SD) | 1.4 (2.3) | 1.3 (3.6) | 0.931 |
| Mean duration of disease, year (SD) | 3.6 (2.7) | 2.9 (4.7) | 0.744 |
| Mean white blood cell count, cell/ml (SD) | 12116.8 (403.7) | 10572.1 (398.9) | 0.367 |
| Mean lymphocyte count, cell/ml (SD) | 3074.2 (2951.6) | 4857.7 (2554.0) | 0.265 |
| Mean IgG, mg/dl (SD) | 1069.0 (296.4) | 636.0 (238.2) | <0.001* |
| Mean IgA, mg/dl (SD) | 231.3 (261.2) | 82.6 (77.6) | 0.001* |
| Mean IgM, mg/dl (SD) | 66.6 (40.1) | 89.3 (33.9) | 0.126 |
| Mean IgE, mg/dl (SD) | 1828.3 (1183.7) | 268.3 (586.8) | <0.001* |
| Mean percentage of CD3 (SD) | 50.5 (7.9) | 66.3 (7.6) | 0.046* |
| Mean percentage of CD4 (SD) | 30.2 (7.5) | 31.6 (13.0) | 0.861 |
| Mean percentage of CD8 (SD) | 18.2 (10.9) | 27.6 (13.5) | 0.344 |
| Mean percentage of CD16/56 (SD) | 7.3 (2.8) | 4.3 (2.0) | 0.152 |
| Mean percentage of CD19 (SD) | 25.7 (7.4) | 20.6 (6.6) | 0.393 |
Oral aphthous ulcers were observed in clinical examination of two cases in Group A (33.3%) and one (1.4%) Group B patient (p=0.016).
Lymphopenia was observed in two patients (33.3%) in Group A and three patients in Group B (4.3%; p=0.023), while eosinophilia was observed in four patients (66.7%) in Group A and 15 cases in Group B (21.7%; p=0.033), respectively. Moreover, thrombocytopenia was observed in two patients (33.3%) in Group A, but none in Group B (p=0.005). Serum levels of IgG, IgA and IgE were significantly higher in Group A (p<0.001, p=0.001 and p<0.001, respectively). Although serum levels of IgM were lower in Group A compared to Group B, this difference was not significant between the two groups (p>0.05, Table 3).
DiscussionIn this study, 8% patients with severe AD were diagnosed with a type of PID (6.6% HIES and 1.3% WAS). WAS and HIES are two types of PIDs that can present with eczematous dermatitis fulfilling the diagnostic criteria of AD.6,19
HIES can be diagnosed by the clinical triad of recurrent staphylococcal skin infections, recurrent sinopulmonary tract infections, and high serum level of IgE (>2000IU/mL).8 Additionally, most of the HIES patients present with papulopustular skin rashes in their neonatal period usually beginning on the face or scalp. These lesions develop to severe eczema that is consistent with the UK Working Party's Diagnostic Criteria for AD in most cases.19,20 There are several reports that showed the co-presence of HIES and AD manifestations.21,22 Differentiation between the HIES and AD is critical due to the importance of the early diagnosis of the HIES since like other PIDs, delay in initiation of an appropriate treatment for these patients can result in a poor prognosis.
The findings of this study suggest some clues for this differentiation, including clinical and laboratory indicators. Similar to our results, previous studies reported some highly specific findings in HIES that can help physicians for this distinguishing such as severe infections, formation of internal abscesses and low counts of TH17 cells.23 From four groups of HIES, the eczema and severe atopy aspects were documented in cases with mutation in signal transducer and activator of transcription 3 (STAT3) and Dedicator of cytokinesis 8 (DOCK8) genes. Although these two entities have indistinguishable clinical features, the history of autosomal dominant pattern in pedigree, history of other allergic disorders, severe cutaneous viral infections, hyperextensible joints, orthopaedic problems as well as the absence of newborn rash and coarse facies have been suggested to favour the clinical diagnosis of DOCK8 deficiency syndrome.5
The most common manifestations of PID patients are recurrent pyogenic infections, including respiratory tract infections and otitis media.11 All of our six PID patients had the history of otitis media and all subjects with HIES (five out of six PID patients) had respiratory infections both with significantly higher frequencies, compared to immunocompetent AD patients.
Pyogenic staphylococcal skin infections are also frequent in immunodeficiency.4 Patients with HIES are susceptible to infections caused by extracellular bacteria and fungi.24,25 The dermatitis occurring in HIES becomes severe due to its colonisation and subsequent infection with Staphylococcus aureus that can be cured by antibiotics. The recurrences can occur in the absence of antibiotic prophylaxis.26 Three of our five patients with HIES had a history of recurrent skin infections that was significantly higher than the number of AD patients with this history. This can be a clue in suspicion to an underlying immune disorder that should be investigated in patients presenting with severe AD.
Concordance with our findings, besides high serum level of IgE, eosinophilia (>800/mm3), is a laboratory finding in diagnostic criteria of HIES.27 Also, eosinophilia can be present in AD and the eosinophil count correlates with the severity of this disease.28 In addition to HIES, there are some other immunodeficiency diseases that can be associated with high levels of serum IgE including WAS, Omenn syndrome, and Comel–Netherton syndrome which can be differentiated by clinical findings and specific genetic defects.29 The serum IgE level of 2000IU/mL is considered as the cut-off point in making the definitive diagnosis of HIES.30 However, the level of IgE is frequently more than 5000IU/mL and in some cases exceeding 100,000IU/mL in this syndrome.8,31 In contrast, serum IgE level in typical AD patients is usually about 1000IU/mL and can be at least approximately 10,000IU/mL.29 However, IgE level can exceed this range in severe forms of AD.22 These data show that the measurement of serum level of IgE cannot help in the differentiation between AD and PIDs with HIES. However, the results of the current study showed that serum IgE level is one of the indicators of HIES identified patients compared to other severe AD subjects.
WAS, characterised by mutation in the WAS gene,32 is diagnosed by the presence of recurrent pyogenic infections and thrombocytopenia in addition to severe eczematous dermatitis.4 Although the low number of population in this type of PID invalidated us from interpreting WAS findings statistically, the records of these patients affect the overall clues for finding PID in severe AD cases. Although WAS is known by the triad of infections’ susceptibility, bleeding tendency and dermatological manifestation, clinical criteria are completed during infancy in only one-third of patients.33 Moreover, based on the classification of Jin et al. intermittent thrombocytopenia could be presented in one subgroup of cases with WAS. Therefore, it could be justified how the patient with WAS initially was diagnosed as atopic dermatitis without a bleeding problem.34
There was a positive family history of PIDs leading to early death in infancy in 6.7% (P4) and family history of death in early infancy without an identified in 33.3% (P2 and P3) of patients with diagnosis of HIES with significant difference with Group B. Therefore, consideration in taking the family history can help in suspicion to the presence of a PID as death in early infancy can be the presentation of these disorders.35
The serum IgA and IgG levels of our studied PID patients were significantly higher than these parameters in AD patients. This could be due to higher occurrence of infections in subjects with PID.
Lymphopenia and thrombocytopenia were detected in our PID patients with significant prevalence, in comparison with Group B patients. These two can be considered as helpful laboratory findings in diagnosis of an immunodeficiency in AD patients.
In summary, among the warning signs of primary immunodeficiency diseases,36 the following were observed in our studied patients who presented with severe AD and had a final diagnosis of a PID: positive family history of a PID, history of recurrent infections, especially respiratory tract and ear infections, recurrent skin infections, and persistent oral aphthous ulcers.
In conclusion, an underlying PID can be the possible reason for poor prognosis and management of some patients with severe AD. In patients with compatible clinical findings of severe AD, an immunological work-up is recommended due to the probable presence of an immune system disorder in selected cases with special indicators. Specific clinical manifestations and laboratory findings raise the suspicion towards PIDs in AD patients, which can result in early diagnosis and proper treatment of these serious disorders.
Ethical disclosuresProtections of human subjectsThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of dataThe authors declare that they have followed the protocols of Tehran University of Medical Sciences on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors have no conflict of interest to declare.
