


To the Editor:
Common variable immunodeficiency (CVID) is a heterogeneous disorder characterized by deficient antibody synthesis. It manifests as recurrent bacterial respiratory infections and an increased incidence of neoplasms – particularly lymphomas, autoimmune processes and granulomatous diseases1–3. We report a clinical case of CVID of special interest in paediatrics due to its initial manifestation as non-Hodgkin lymphoma.
A four-year-old boy presented with headache, asthenia and anorexia for the previous 15 days. The oncological history among his grandparents revealed leukaemia and stomach, lung, and testicle cancer. The paternal grandmother suffered diabetes mellitus and chronic arthritis; the maternal grandmother presented hypothyroidism. There were no personal disease antecedents of interest other than very intense varicella at 18 months of age, and repeat bronchitis up to three years of age. Vaccinations were correct and well tolerated.
Physical examination revealed a poor general condition, 2/6 systolic murmur, oral candidiasis, submandibular adenopathies and abdominal distension with hard and painful 4 cm hepatomegaly associated with nodular areas. The neurological exploration proved normal.
The initial complementary explorations revealed intense anaemia in the blood tests (haemoglobin 9.7 mg/dl, haematocrit 34 %), important lactate dehydrogenase elevation (LDH 1936 U/l), abdominal ultrasound findings in the form of diffuse hepatomegaly of both lobes and multiple nodular images compatible with liver metastasis. The brain CT scan in turn showed oedema and a hypodense nodular image containing calcifications in the left cerebellar hemisphere, suggestive of metastasis.
The initial diagnostic impression was lymphoma, in view of the age of the patient, the important family antecedents of oncological disease, and the aggressivity of the clinical condition.
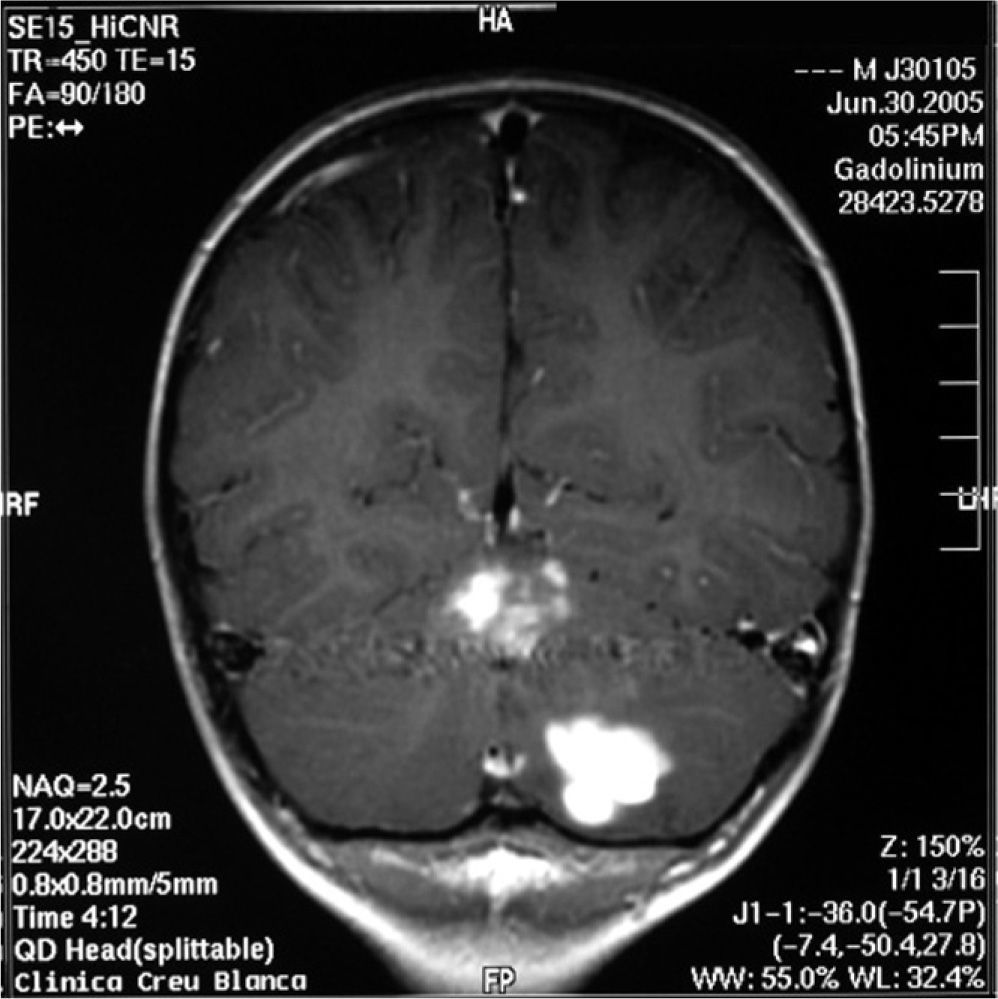
The study was continued with brain and spinal MRI, which revealed expansive lesions in the left hemicerebellum with nodular contrast uptake and an oedematous halo, compatible with lymphoma (fig. 1).

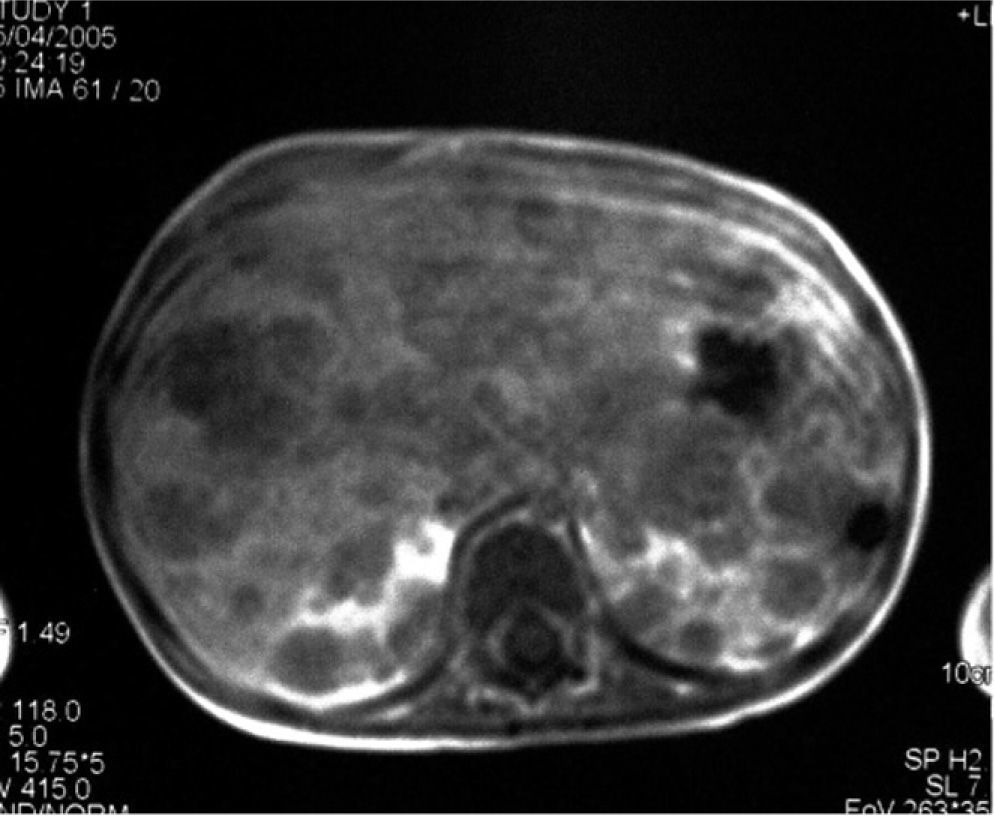
There was no spinal involvement. The abdominal CT scan identified nodular lesions in the liver and kidneys, as well as mesenteric adenopathies (fig. 2). The bone marrow biopsy confirmed the existence of a blast cell infiltrate with an immunophenotype corresponding to leukaemia/B lymphoma, with FAB L3 morphology.

At that point, the immune study revealed a decrease in all immunoglobulins, with normal lymphoid populations (IgG 2810 mg/l, IgM 180 mg/l, IgA 220 mg/l, lymphocytes 1697/ mm3, B lymphocytes 350/mm3, T lymphocytes 1273/mm3 CD8 593/mm3; CD4 680/mm3), and natural killer cells 74/ mm3. Of note was the absence of anti-rubella IgG antibodies and of anti-tetanus IgG and IgM antibodies, despite previous vaccination against those diseases. The titres of those antibodies were measured three times after double checking that the patient was correctly vaccinated and re-vaccinated against measles, rubella, mumps, diphtheria, tetanus and whooping cough. In all three measurements the aforementioned antibodies were absent.
With the diagnosis of leukaemia / Burkitt lymphoma, combined treatment was provided with systemic and intrathecal chemotherapy. The cerebrospinal fluid repeatedly proved normal. The patient showed complete bone marrow and systemic remission, though without improvement of the cerebellar lesions. Radiotherapy of the central nervous system was provided, with persistence of the cerebellar lesions. In this context, the difference in behaviour between the nervous system lesions and the rest of the body lesions must be stressed — with no response to treatment on the part of the lesions in the central nervous system. A biopsy of the cerebellar lesions revealed a chronic infíltrate compatible with reactive changes in response to oncological treatment, with no evidence of neoplastic disease.
During follow-up, the blood tests repeatedly indicated low immunoglobulin levels. The patient was therefore referred to our Section of Immunology. Eighteen months after diagnosis of the lymphoma and six months after the completion of chemotherapy, the patient showed diminished immunoglobulin levels and CD4+ lymphocyte counts (IgG 2902 mg/l, IgM 347 mg/l, IgA 471 mg/l, CD4+ 680/mm3), with the absence of anti-rubella and anti-tetanus IgG antibodies. A review of the case history showed that laboratory tests requested by the paediatrician usually attending the patient two years prior to the diagnosis of lymphoma for the evaluation of recurrent bronchitis already revealed low IgG and IgA values (IgG 5530 mg/l, IgA 210 mg/l, IgM 670 mg/l). Possible Immunoglobulin losses through the digestive and urinary systems were ruled out as there was neither chronic diarrhoea, nor malabsorption syndrome or nephropathy.
This case corresponded to a 6-year-old boy with a history of Burkitt lymphoma, persistent hypogammaglobulinemia, a lack of response to vaccination, and a quantitative defect in cellular immunity. At that point common variable immunodefíciency was diagnosed which had already been present before the lymphoma. Treatment was started with intravenous gammaglobulin (6 g/month), with a good course, normalisation of immunoglobulin values, and no infections. The cerebellar lesions remain stable in the imaging controls, and the patient is asymptomatic.
Common variable immunodeficiency (CVID) is a primary immune defíciency characterized by the existence of defícient antibody synthesis. The clinical and immunological profíle of the disorder is very heterogeneous. The defects include anomalies in B lymphocyte survival, a reduction in CD27+ memory B cells, failure in immunoglobulin isotype change to IgA and IgG, and defective B lymphocyte activation1,4. In some cases anomalies in T lymphocyte function and number may be observed1,4.
The estimated incidence of CVID is from 1:10,000 to 1:50,0002. The disorder is related to selective IgA deficit, which is a much more common and often asymptomatic condition. Both disorders may coincide in the same family and even in the same individual, and some patients with IgA deficit evolve towards CVID 1. A common genetic defect has been postulated in these two diseases (mutations in genes of the major histocompatibility complex, IGAD1)1,4.
CVID can manifest at any age, though there are two peaks in incidence: between 5–10 years of age, and in young adults between 20–30 years of age2,3. The most frequent form of presentation is in the form of recurrent respiratory infections: pneumonias, bronchitis, otitis, sinusitis and conjunctivitis. The most common causal pathogens are encapsulated bacteria: Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis1,2. Inadequate management of such respiratory infections may lead to the development of bronchiectasis.
Digestive infections are also frequent. In this context, Giardia lamblia is the most common cause of diarrhoea, although other pathogens such as Salmonella, Shigella and Campylobacter can also be implicated2,3.
Viral infections do not usually pose a problem, although some patients suffer recurrent infections produced by herpes simplex (HSV-1) or herpes zoster (HZV)2.
A very small number of patients may present infections produced by mycobacteria, fungi and Pneumocystis carinii2.
Twenty percent of all patients develop autoimmune diseases the most common being idiopathic thrombocytopenic purpura and autoimmune haemolytic anaemia 3. CVID is also associated to rheumatological problems such as rheumatoid arthritis, systemic lupus erythematosus and vasculitis 1–3. Autoimmune phenomena may constitute the first manifestation of the disease2.
Some patients develop inflammatory bowel disease, Crohn's disease or ulcerative colitis in early or late stages of the disease1–3.
In 10 % of cases granulomas are identified the lungs being the most commonly affected location, though granulomas can also be found in the skin, intestine, liver, or with a generalised distribution simulating sarcoidosis1,2.
A very common finding is non-malignant lymphoproliferation in the form of splenomegaly, adenopathies and gastrointestinal lymphoid nodular hyperplasia2.
Affected patients are at an increased risk of developing neoplasms, such as extranodal B cell lymphomas and gastric cancer1,2.
Recently, genetic defects in ICOS, CD19, TACI and BAFF-R have been identified; all these molecules are implicated in the regulation of B lymphocyte activation and terminal differentiation 4,5. TACI and BAFF-R are essential for B cell homeostasis, and play an important role in immunoglobulin isotype change. CD19 deficiency underlines the importance of the B lymphocyte receptor signalling pathway, while ICOS deficiency illustrates the importance of B:T cell interaction in coordinating an effective secondary immune response4,5. These defects account for 10–15 % of all cases of CVID. New genetic defects in relation to the activation of B lymphocytes are likely to be identified, and in future CVID could be classified according to the different molecular defects involved.
We consider the present case to be of interest because it reflects the clinical and immunological heterogeneity of this disease. The patient was a four-year-old boy initially manifesting with non-Hodgkin lymphoma, followed one year later by the diagnosis of CVID due to the existence of persistent hypogammaglobulinemia that had already been present before diagnosis of the lymphoma. The important familial oncological and autoimmune antecedents of this patient make this case of particular note.
