


X-linked agammaglobulinaemia (XLA) is a genetic disorder characterised by a defect in the generation of mature B cells, lack of antibodies production, and susceptibility to recurrent bacterial infections. Understanding of the risk factors responsible for morbidity and mortality in these patients can help in a better management of this disorder. However, there is a lack of specific studies in the literature regarding the morbidity and mortality of XLA patients. This study is designed to evaluate morbidities and mortality and survival rates in Iranian patients with XLA diagnosis during the past 20 years.
MethodsWe have registered the clinical data of the XLA patients and followed them up until 2010. At the time of diagnosis, a four-page questionnaire including complete medical information was filled out for all patients. Follow-up information was gathered either by reviewing the patients’ hospital records or regularly visiting the patients.
ResultsAmong 41 patients, 26.8% died during the follow up period. All of the complications before the initiation of treatment such as pneumonia, otitis media and diarrhoea were reduced after immunoglobulin replacement, except sinusitis and conjunctivitis. There were significant associations between some immunological and clinical characteristics such as lymphocyte subsets, consanguinity marriage and mortality.
ConclusionDespite recent advances in the treatment of XLA, these patients still suffer from severe complications. Associations between poor prognosis and clinical and some immunological characteristics of the patients may help physicians to select poor prognoses patients at higher risk of mortality to develop prevention strategies for them.
X-linked agammaglobulinaemia (XLA) is an X-linked recessive disorder of the immune system characterised by a defect in the generation of mature B cells, very low serum levels of all immunoglobulin isotypes, and the lack of specific antibody production.1 XLA is caused by mutations in the gene responsible for the production of B-lymphocyte tyrosine kinase (BTK), leading to a defective humoral immune response.2–4 These patients are susceptible to recurrent bacterial infections, mainly of the respiratory and gastrointestinal tracts for which lifelong intravenous immunoglobulin (IVIG) replacement is indicated to prevent these complications.5–7
Over the past decades, the mortality and morbidity of XLA patients have been significantly reduced by the use of IVIG replacement therapy. However, its usage is only in the management of the disease instead of its cure, and is associated with several long-term complications and its beneficial effect has been more pronounced for the control of systemic rather than of mucosal infections.8–11 The major clinical complications in these patients are due to recurrent encapsulated bacterial infections, such as the pneumococcus and Haemophilus influenzae (either encapsulated or unencapsulated), as well as parasites (e.g. Giardia lamblia) and severe viral infections (e.g. enteroviruses).6,7,12,13 Respiratory infections are still the major cause of morbidity and mortality in XLA patients.13
Understanding of the risk factors responsible for morbidity and mortality in this group of patients can guide physicians into a better follow-up and management process. In addition, such information may potentially direct the clinician towards selecting XLA patients who are at higher risks of death and complications, for more supportive management and prevention strategies. The purpose of this study is to evaluate morbidities, mortality and survival rates in Iranian patients with XLA, during the past 20 years (1990–2010).
Materials and methodsPatientsChildren's Medical Centre Hospital serves as a referral centre for patients with a known or suspected immunodeficiency disease in Iran.14 In this study, we have selected the clinical data of all the registered patients with XLA diagnosed and followed up between 1990 and 2010. XLA was diagnosed according to criteria introduced by the European Society for Immunodeficiency (ESID) and the Pan-American Group for Immunodeficiency (PAGID).15,16 Other causes of recurrent infections were rolled out in studied patients including atopy, anatomical defect and secondary immunodeficiency. Informed consent was obtained from all patients or their legal warden. The process of this study was approved by the Ethics Committee of the Tehran University of Medical Sciences.
MethodsAt the time of diagnosis, a questionnaire was completed for all patients that contained the patient's complete demographic information, date of birth, first clinical presentation, age at onset of symptoms, age at the time of diagnosis, history of recurrent and chronic infections, autoimmunity and malignancy and other complications.14 Immunoglobulin G (IgG), A (IgA), M (IgM) and CD markers were measured in all patients at the time of diagnosis and later at the follow-up visits using the standard immunochemical assays.1,4,15 For each patient, the percentages of circulating T (CD3+, CD3+CD4+, CD3+CD8+ lymphocyte) and B lymphocytes (CD19+ lymphocytes) were evaluated at the time of enrolment by FACS analysis. All patients received standard dosage of IVIG and IgG levels were evaluated to monitor therapy based on the recommended values. Follow-up visits were documented either by reviewing the patients’ hospital records or interviewing the patients or their guardians. Diagnostic delay was considered as the time between the onset of symptoms and the diagnosis. For those who had died, the cause of death was determined by reviewing the death certificate, autopsy report, and/or by contacting the attending physician.
Statistical methodsData analysis was performed using the SPSS statistical software package version 16.0. One-sample Kolmogrov–Smirnov test estimated whether data were normally distributed. Parametric and non-parametric analyses were performed based on the finding of this evaluation. Probabilities of survival after diagnosis of XLA were estimated from Kaplan–Meier life method. A P-value lower than 0.05 was considered statistically significant.
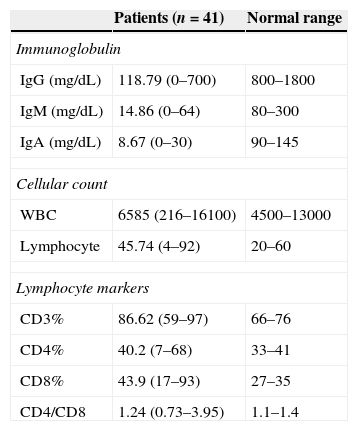
ResultsPatient characteristicsForty-one male patients were entered into the study. At the final time of the study the mean (SD) age of the patients was 18.3 (8.5) years. The mean (SD) age of the patients at the onset of symptoms and at the time diagnosis was 1.5 (1.3) years and 4.9 (3.6) years, respectively. The mean (SD) diagnostic delay was 3.4 (3.1) years. Parents of 10 patients (24.3%) were relative and 26 patients (63.4%) had a positive family history of primary immunodeficiency (PID). Consanguinity was more observed in patients with a positive family history of PID (P<0.05). Serum immunoglobulin levels and immunophenotyping of peripheral blood lymphocytes are summarised in Table 1.
Immunological characteristics of the study population at the time of diagnosis.
| Patients (n=41) | Normal range | |
|---|---|---|
| Immunoglobulin | ||
| IgG (mg/dL) | 118.79 (0–700) | 800–1800 |
| IgM (mg/dL) | 14.86 (0–64) | 80–300 |
| IgA (mg/dL) | 8.67 (0–30) | 90–145 |
| Cellular count | ||
| WBC | 6585 (216–16100) | 4500–13000 |
| Lymphocyte | 45.74 (4–92) | 20–60 |
| Lymphocyte markers | ||
| CD3% | 86.62 (59–97) | 66–76 |
| CD4% | 40.2 (7–68) | 33–41 |
| CD8% | 43.9 (17–93) | 27–35 |
| CD4/CD8 | 1.24 (0.73–3.95) | 1.1–1.4 |
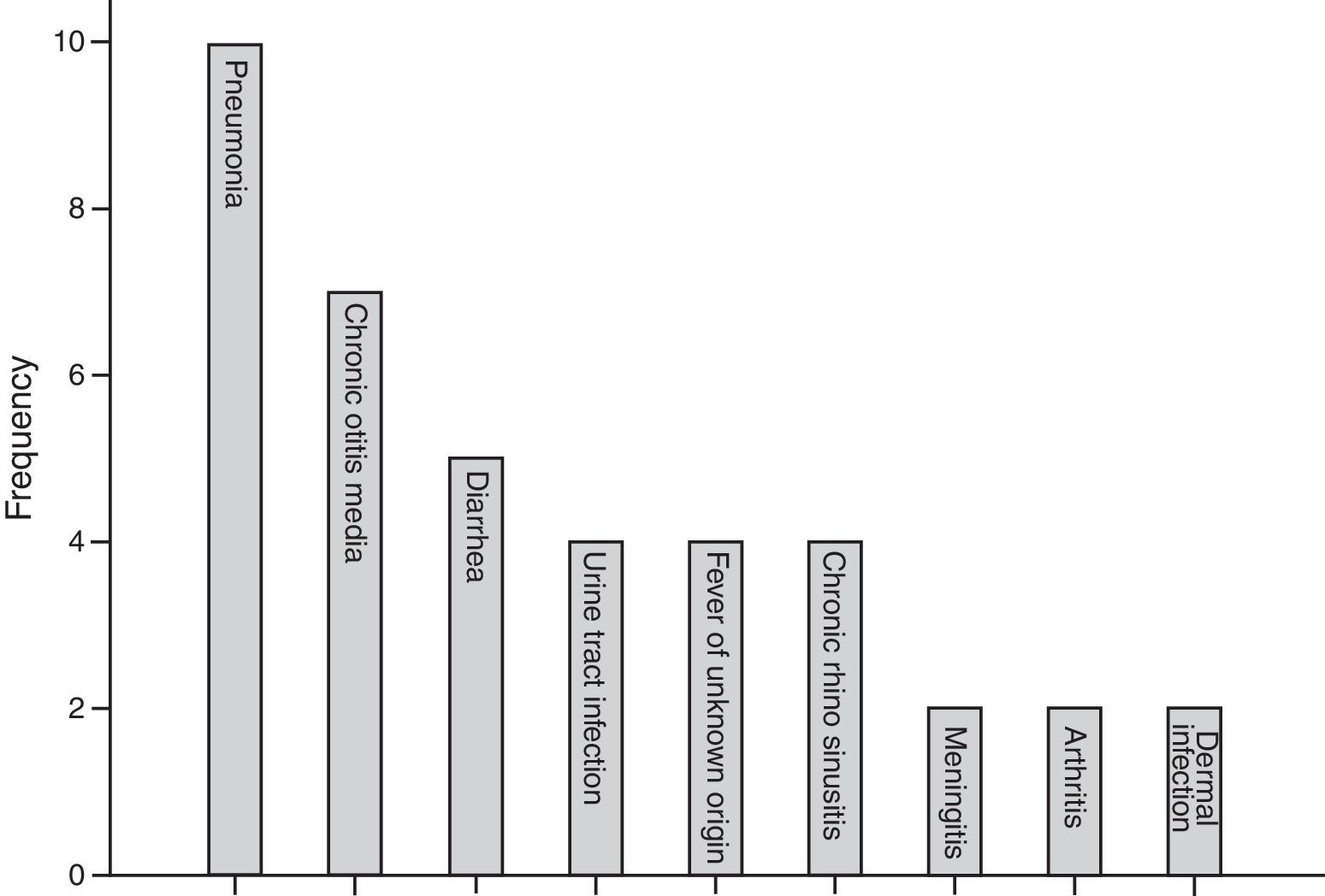
The most common first presentation was pneumonia in 10 patients, chronic otitis media in seven (17.0%), and diarrhoea in five (12.1%). Three patients (7%) also suffered from failure to thrive. Fig. 1 summarises the most prevalent first manifestation of XLA patients. The main complication of the patients before and after treatment with IVIG is also investigated. The most common manifestations before treatment were pneumonia in 31.7% of patients and 21.9% of chronic otitis media, while the most common manifestation after treatment was sinusitis in 34.1% of patients. Vaccinations and their complications were checked for all patients. Eight (20%) out of 41 patients received complete oral polio vaccination which resulted in paresis in two of polio receiving patients (25%).
Mortality
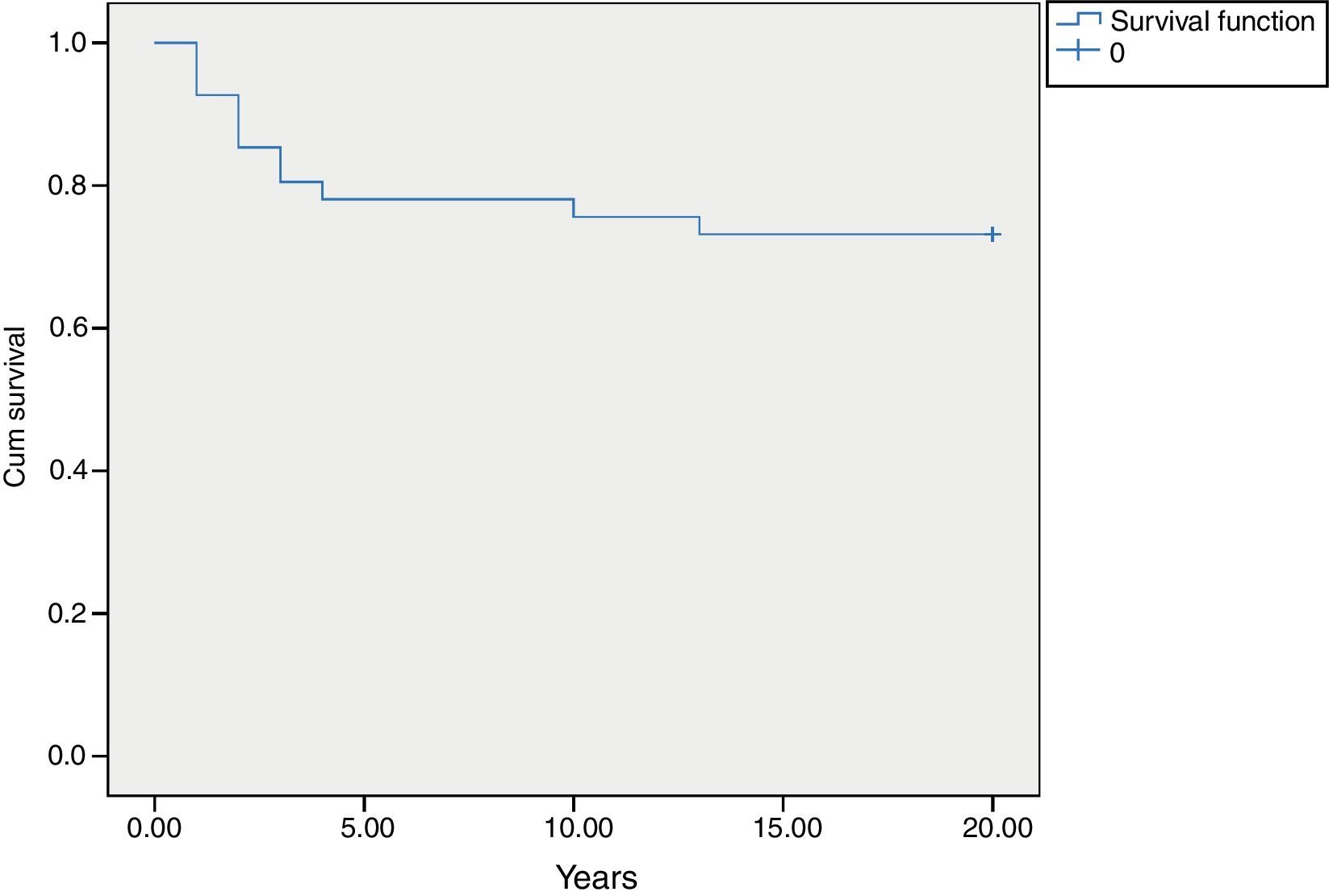
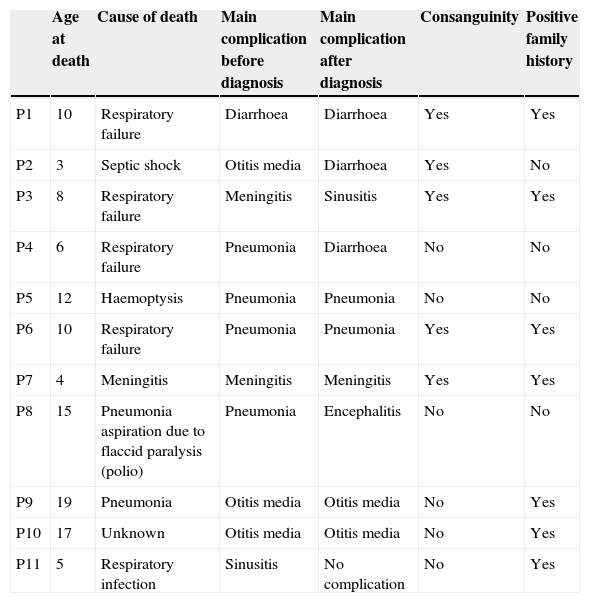
Among 41 patients, 11 patients (26.8%) died during the follow up period (Table 2). Twenty-four patients (58.5%) were known to be alive and the condition of six patients (14.6%) was unknown. Hospitalisation rate for dead patients was significantly higher than alive patients (2.1±1.02 vs. 0.87±0.52; P<0.05). Statistical analysis did not show a significant association between the diagnosis delay and mortality (P=0.8). Post diagnosis survival was estimated as 78% for the first five years (Fig. 2). The survival rate was not shown to be influenced by delayed diagnosis, type of complications, serum levels of immunoglobulin and lymphocyte or WBC count at the time of diagnosis. CD4 cells and CD4/CD8 were higher comparing to dead patients (P=0.013 and P=0.01 respectively). But oppositely, CD8 cells were significantly higher in alive patients (P<0.01).
Clinical characteristics of dead patients.
| Age at death | Cause of death | Main complication before diagnosis | Main complication after diagnosis | Consanguinity | Positive family history | |
|---|---|---|---|---|---|---|
| P1 | 10 | Respiratory failure | Diarrhoea | Diarrhoea | Yes | Yes |
| P2 | 3 | Septic shock | Otitis media | Diarrhoea | Yes | No |
| P3 | 8 | Respiratory failure | Meningitis | Sinusitis | Yes | Yes |
| P4 | 6 | Respiratory failure | Pneumonia | Diarrhoea | No | No |
| P5 | 12 | Haemoptysis | Pneumonia | Pneumonia | No | No |
| P6 | 10 | Respiratory failure | Pneumonia | Pneumonia | Yes | Yes |
| P7 | 4 | Meningitis | Meningitis | Meningitis | Yes | Yes |
| P8 | 15 | Pneumonia aspiration due to flaccid paralysis (polio) | Pneumonia | Encephalitis | No | No |
| P9 | 19 | Pneumonia | Otitis media | Otitis media | No | Yes |
| P10 | 17 | Unknown | Otitis media | Otitis media | No | Yes |
| P11 | 5 | Respiratory infection | Sinusitis | No complication | No | Yes |

There are several diversities between registries of XLA patients worldwide. In an Argentinean group of patients with XLA the mean age of onset was one year.16 But in another study done by Plebani et al. in Italian cases the mean age at the time of onset was two and at diagnosis was 3.5 years.13 The time of onset usually begins after the first few months of life due to the presence of acquired maternal immunoglobulin in the blood. Typically, patients are affected by recurrent bacterial infections particularly otitis, conjunctivitis, pneumonitis, diarrhoea, and skin infections in the first two years of life and are diagnosed as XLA before their fifth year of life.12,13 The diagnosis delay in our series was somewhat higher than in other studies.16 This considerable delay shows the poor awareness of this condition among general practitioners and paediatricians in our country.17,18
The most common chief complaints in our patients were pneumonia, otitis media and diarrhoea. Based on previous studies, recurrent otitis is the most common infection before the diagnosis. Conjunctivitis, sinopulmonary infections, diarrhoea, and skin infections were also frequently reported19,20. Early detection of antibody deficiency syndromes is vital to prevent chronic infections such as tissue damage. It has been shown that immunological defects have an important role in recurrent ear and sinus infections, so the immune system of patients with these manifestations should be evaluated. Although respiratory and gastrointestinal tract infections are the most common infections seen in patients with XLA, other infections such as meningitis and septic arthritis also occur.21 Among our patients, two patients had meningitis, two had septic arthritis and one had dermal manifestations. After treatment with IVIG, the distribution of main complications changed in our patients. Dominant manifestations before treatment such as otitis media were replaced by sinusitis.
Chronic sinusitis was a common long-term complication in previous studies which is in agreement with the result of our study.13 Treatment of sinusitis is still challenging, since both medical treatment and surgical treatment of sinusitis are not curative in these patients and can at best provide only temporary improvement.5,13,22,23 Because the protection at mucosal surfaces is normally provided by secretory IgA and IgM antibodies, treatment with high-dose IVIG would not be so useful in the prevention of sinus infections.23–25
In our patient group, two patients have developed paresis due to the polio vaccination; another two patients had gastroenteritis before receiving IVIG but this complication was managed after receiving the treatment. Before the mid-1980s, 5–10% of patients with XLA developed vaccine-associated polio after vaccination with the live attenuated oral polio vaccine. But after introduction of IVIG as a treatment option for patients with XLA, vaccine-associated enteroviral infection has markedly decreased. Another reason could be the use of inactivated polio vaccine in developed countries. However, some patients still develop enteroviral encephalitis and paresis.26,27
In the families with a pervious history of primary immunodeficiency, the consanguine marriage of the parents was a dominant feature. This emphasizes the probability of coincidence of both XLA and an autosomal recessive immunodeficiency in these patients which should be investigated in the future study.
The rate of mortality in our patients was 26.8%. This mortality rate was similar to that reported in the two earlier large surveys,19,21 but much higher than that reported in more recent surveys on XLA patients.5,13 The prognosis has improved markedly for patients with XLA in the last two decades28 as a result of earlier diagnosis, more liberal use of antibiotics, and treatment with IVIG. Most individuals can experience a near normal life.5 The main cause of mortality in our patients was respiratory failure due to the chronic respiratory infections which suggests that, despite appropriate serum IgG level of patients during follow-up period, chronic lung disease (CLD) remains the most dangerous complication in XLA patients. There were significant relationships between some immunological parameters of our patients at the time of diagnosis and prognosis.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the responsible clinical research ethics committee and in accordance with those of the world medical association and the Helsinki declaration.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors have no conflict of interest to declare.
