


The Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive immunodeficiency disorder, caused by mutations in the WAS protein (WASP) gene and characterised by thrombocytopenia, small platelets, eczema, and recurrent infections associated with increased risk of autoimmunity and malignancy disorders. The gene for WAS has been mapped to the short arm of the X chromosome at Xp 11.22-23 and early detection of patients and diagnosis of new mutation might reduce related complications and increase their life expectancy.
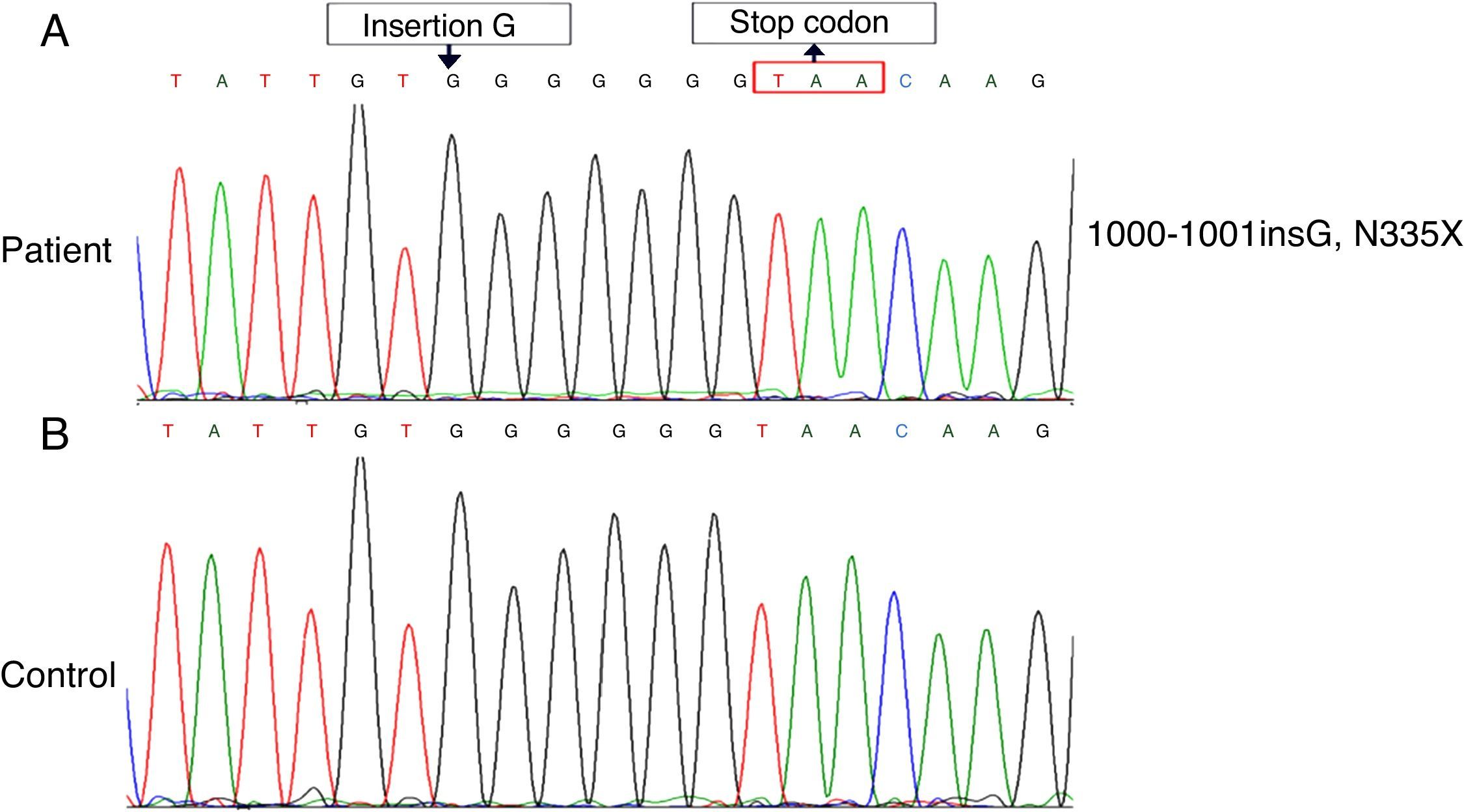
Method and resultWe found a novel mutation by sequence analysis of genomic DNA coding of a 9-month old boy suffering from WAS. The mutation was insertion G in exon 10 of WASP gene. The consequence of the G insertion is a premature stop immediately at amino acid 335 (N335X or p.G334GfsX1) and truncated protein.
ConclusionThe mutation analysis is helpful for the diagnosis of WAS patients and also expanding the spectrum of WASP mutations for carrier detection and prenatal diagnosis.
The Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive immunodeficiency disorder characterised by eczema, thrombocytopenia with small platelets, recurrent infections and high risk for autoimmunity and malignancy.1,2 In WAS patients, thrombocytopenia is an exclusive health concern which is not usually found in other immunodeficiency disorders.
Although malignancy of the reticuloendothelial system and autoimmune disorders such as vasculitis and autoimmune haemolytic anaemia have been associated to WAS,3,4 the most important causes of death in WAS patients has been attributed to infections, bleeding or malignancy.5,6 A functional deficit of WAS gene mutation is also associated with reduced number and function of T lymphocytes and B lymphocytes and natural killer cell dysfunction.7
Early diagnosis of WAS is an important factor in a better prognosis and management in treatment.
Since haematopoietic stem cell transplantation (HSCT) has become the mainstay of treatment for the Wiskott–Aldrich syndrome (WAS), its severe complication including GVHD and infection diseases has restricted HSCT.8 Some recent studies have revealed HSC gene therapy as a potential treatment in WAS patients.9,10
In this regard, Moratto et al. who analysed long-term outcome of HSCT in WAS patients have revealed an overall survival of 84.0% for those who received HSCT.11 Abina et al. also supported the feasibility of HSCT in WAS patients recently.12
A previous study has shown lentiviral gene therapy as a promising treatment for WAS when matched donors are unavailable.13
WAS is a mutation in a gene known as WASP, located on the short arm of the X chromosome (Xp11.4-p11.21) which consists of 12 exons and encodes 502 amino acids.14–16
WASP is expressed solely in all haematopoietic cell-linages and has five functional domains linked to actin polymerisation and intracellular signal transduction. Abnormal or absent WASP protein caused defective platelet formation as well as dysfunctions in innate, humoral and cellular immunity.17,18
At least 300 unique mutations have been identified in the WASP gene, through all 12 exons. WASP mutations consist of missense and nonsense, insertions, deletions, splice site mutations and complex mutations.19,20 The missense mutations, as the most common mutations in patients with WAS, are located in the first four exons in WIP-binding domain.21 Usually, splice site mutations are described in introns 6–10; while deletions and insertions are reported through the whole WASP gene.19,20,22 Gain of function mutations in GTPase-binding domain (GBD) of WASP caused X-linked congenital neutropenia,23 while loss of function mutations leads to WAS and X-linked thrombocytopenia (XLT).20,24,25
Despite various mutation in WASP gene among WAS patients, data regarding relationship of phenotype and genotype is sparse. A previous study which examined the genotype correlation with phenotype in 24 WAS patients has revealed a missense mutations in both WAS phenotype in both milder form and severe disease.26
In this study, we described an Iranian boy with WAS phenotype with a novel WASP mutation.
Case reportA nine-month old boy was referred to our centre with severe eczema since two months after birth. At four months of age, the patient had streaks of blood covering faeces. He had some minor epistaxis but did not have any heavy bleeding. The patient was suffering from petechia and purpura in his trunk and extremities as a result of thrombocytopenia. The patient did not have any history of recurrent infections including recurrent diarrhoea, rhinitis, sinusitis or pneumonia. He was from non-consanguineous parents. He had six healthy maternal aunts, but his only maternal uncle died at childhood. At the time of referral, the white blood cell count was 8230 (103/μl); haemoglobin 10.5 (g/dl) and platelets (Plt) 23,000(/μl). The differential showed 49.2% lymphocytes and 21.7% neutrophils. The serum levels of immunoglobulins for IgG, IgA, IgM and IgE were 1161mg/dL, 121mg/dL, 97mg/dL, and 2IU/mL, respectively. Repeated CBCs were also compatible with severe thrombocytopenia (20,000/μl and 10,000/μl). Since the patient had thrombocytopenia with small platelets and mild, transient eczema without infections received a score of 2 in Zhu score.
The clinical diagnosis of WAS was made for the patient. By the sequence analysis of all exons, a novel insertion mutation, c.1000-1001insG (N335X), reported that led to a stop codon at amino acid 335 (Fig. 1). The patient is candidate for Allogeneic HSCT from his brother.
Discussion Normal. (B) The mutation, 1000–1001 ins (G), which lead to a frameshift (p.G334GfsX1), resulting in a premature termination codon at amino acid 335 (N335X).")
Wiskott–Aldrich syndrome is a rare immune system disorder caused by mutation in a gene on the short arm of chromosome X, which was termed Wiskott–Aldrich syndrome protein gene (WASP).27 The WASP gene codes instructions for making a protein called WASP which is 502 amino acid long and is mainly expressed in haematopoietic cells. WASP is involved in activating actin polymerisation by binding to the Arp2/3 complex.28 In T-cells, Wasp activation via T-cell receptor signalling pathways allows actin cytoskeleton interaction which is responsible for immunological synapse.29
Lack of functional WASP signalling impairs the function of the actin cytoskeleton in developing blood cells.30 White blood cells that lack WASP have less ability to respond to their foreign invaders and form immune synapses causing many of the immune problems related to Wiskott–Aldrich syndrome. Furthermore, loss of functional WASP in platelets disrupts their development, leading to reduced size and early cell death.31
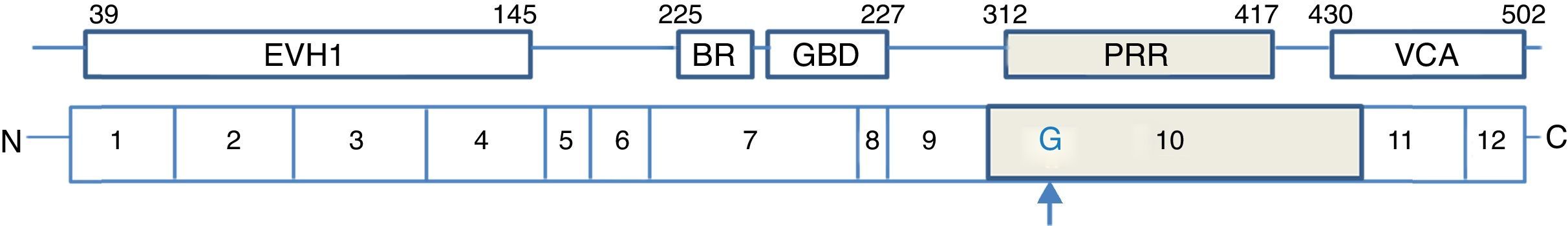
WASP contains several domains, including an Enabled (Ena) and vasodilator-stimulated phosphoprotein (VASP) family, also referred to as a WASP homology domain 1 (WH1), a short basic region (BR), GTPase-binding domain (GBD), a proline-rich region with SH3 binding motifs in N-terminal WASP protein. The C-terminal protein of WASP contains a VCA region, a G-actin-binding verprolin homology (V) domain, a cofilin homology (C) domain and a C-terminal acidic (A) segment (Fig. 2). The central acidic region is involved in activating the actin-related protein (Arp)2/3 complex that lead to reorganisation of the actin cytoskeleton.32,33
 in exon 10 in PRR.")
In inactive WASP, an auto inhibited conformation, the verpoline/acidic regions interacts with the GTPase-binding domain. Activation of WASP carries out by binding of Cdc42 to the GBD domain, phosphorylation of WASP-interacting protein (WIP), and binding of SH3 domains containing proteins (such as Nck) to the proline-rich region. The active form of WASP causes actin polymerisation. The N-terminal region of WASP interacts with WIP, binding to the EVH1 domain and stabilising WASP.34 The WIP/WASP complex is disrupted by phosphorylation of WIP that is allowed to activate WASP by Cdc42.35
The detection of mutation of WASP was performed by direct sequence analysis of all exons. In this study, we report a unique case of WAS with a novel mutation, insertion of nucleotide (G) in exon 10 (c.1000-1001 ins G), which led to a frameshift (p.G334GfsX1), resulting in a premature termination codon at amino acid 335 (N335X) in the PRR (proline-rich region) of WASP (Fig. 2).
A proline-rich region in exon 10 has been shown to be necessary for efficient employment of WASP to the immunological synapse in T cells and was required for optimal actin polymerisation activity.36 This sequence presents binding sites for SH3-domain containing proteins that include the adaptor proteins P47nck and several protein tyrosine kinases including Btk which directly activate WASP through tyrosine phosphorylation.37–39
As mentioned above, multiple SH3 domains of Nck are required for activation of WASP.
Therefore, truncated WASP protein with defective in PRR caused inactive protein. Furthermore, in this mutation due to lack of VCA region, the shortened WASP cannot relate with Arp2/3 complex, and lead to disrupt actin polymerisation.
A previous study has described dual mutation in the same gene area (exon 10) in a patient with WAS. One mutation was a one-base insertion and the other was a one-base deletion (G−) which led to frame shift.40
The absence of protein or truncated protein is typically related to severe clinical affected patients. However, insertion of nucleotide (G) in exon 10 reduces a few part of WASP after the GBD domain and leaves N-terminal of WASP. So it would be presumed that the truncated WASP can combine with WIP because a WH1 domain still remains intact for WIP binding. Therefore, truncated WASP can be relatively stable and protects from destroying.
In summary, we report a novel mutation N335X or p.G334GfsX1 of the WASP gene in a boy with classic WAS. The mutation resulted in a truncated WASP with manifesting of clinical WAS.
The sequence analysis of genomic DNA coding of the WASP suggests diagnostic detections in patients with WAS and in carrier detection and prenatal diagnosis and also offer expanding the spectrum of WASP mutations for Wiskott–Aldrich syndrome. Other methods such as protein expression analysis of WASP for diagnosis of new mutations should always be attached with direct mutation analysis to compare variation in phenotypes of affected patients. Therefore the western blot method can be done in future approaches.
Authors contributionsAll authors have been contributed significantly to the work, have read the manuscript, attest to the validity and agree to its submission.
Ethical disclosuresProtection of human subjects and animals in researchThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the text.
Conflict of interestThe authors have no conflict of interest to declare.
Funding SourceNone.
