


INTRODUCCION
Los corticoides, casi 50 años después de su introducción, siguen siendo uno de los pilares principales de la terapéutica dermatológica. Sin embargo, su uso está limitado por sus efectos secundarios locales y sistémicos. Existe una enorme demanda de medicamentos con capacidad antiinflamatoria que no pertenezcan al grupo de los corticoides que sean útiles en distintas dermatosis inflamatorias.
En los últimos años el arsenal terapéutico para la dermatitis atópica se ha incrementado con 2 grupos diferentes de medicamentos: los inmunomoduladores tópicos y los inhibidores de los leucotrienos. Ambos grupos de medicamentos son novedosos y probablemente veamos aparecer nuevos compuestos pertenecientes a este tipo de fármacos en los próximos años.
La experiencia acumulada en los distintos trabajos parece indicar que el tacrolimus (y con menor cantidad de evidencia) es un tratamiento seguro y eficaz de la dermatitis atópica. El número y la calidad de la evidencia aportada por los estudios de tratamiento con inhibidores de los leucotrienos es mucho menor, pero también parecen un conjunto de sustancias sobre las que es necesario seguir investigando.
El objetivo del trabajo es revisar lo publicado hasta la fecha en literatura inglesa y española sobre el papel de los inmunomoduladores tópicos y los inhibidores de los leucotrienos en la dermatitis atópica.
INMUNOMODULADORES TOPICOS
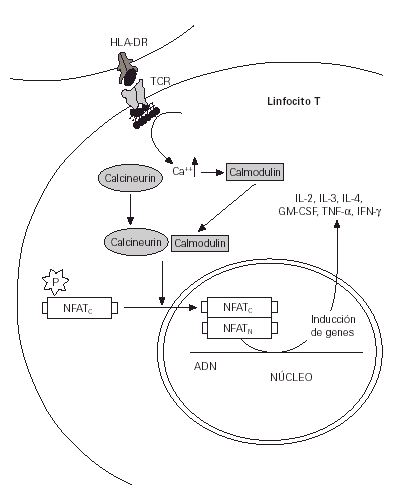
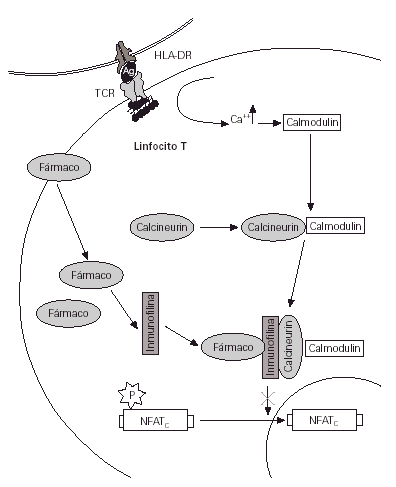
Entre las alternativas a los corticoides se encuentran los inmunomoduladores, y de todos ellos los antibióticos macrólidos destacan como los más prometedores (1). El tacrolimus (FK 506) y el pimecrolimus (ASM 981) pertenecen a este grupo de sustancias que poseen una elevada capacidad de inhibición de la expresión de diferentes genes que participan en el proceso de activación de los linfocitos T (2) (figs. 1 y 2).
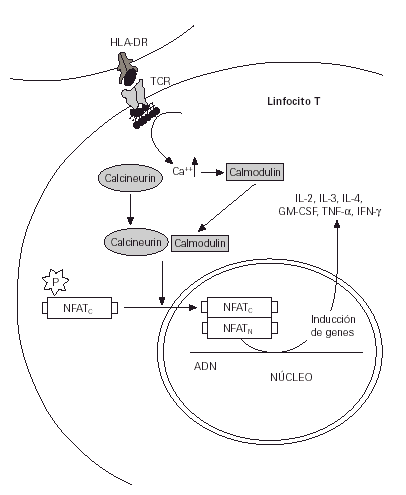
Figura 1.--Mecanismo normal de estimulación de los linfocitos T a partir de la presentación de antígenos.
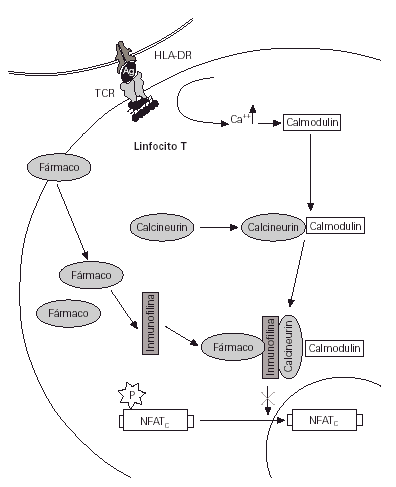
Figura 2.--La inhibición de la acción de la calcineurina por parte del inmunomodulador (tacrolimus o pimecrolimus) impide la defosforilación y el paso del factor activador citosólico al núcleo. Con ello algunos genes se mantienen reprimidos y finalmente no hay nueva síntesis de citocinas.
La ciclosporina es otro inmunosupresor con acción similar. Sin embargo, a diferencia del tacrolimus y el pimecrolimus, no es activa por vía tópica, lo que limita su empleo dermatológico debido a los potenciales efectos secundarios de su uso sistémico (3). El tacrolimus ha demostrado actividad en su empleo tópico en ensayos clínicos a doble ciego en humanos (4-12) y en animales de experimentación (13-15). Aunque el número de ensayos es menor, también se han publicado datos sobre la eficacia de la SDZ ASM 981 en dermatitis atópica (16, 17).
La situación de ambos macrólidos es diferente. El tacrolimus fue comercializado en Japón en 1999 y en EE.UU. a finales de 2000. Se espera que sea comercializado a lo largo de 2002 en la Unión Europea. El pimecrolimus todavía no ha sido comercializado en ningún país del mundo y se haya en estadio III de investigación clínica.
MECANISMOS DE ACCION DE LOS INMUNOMODULADORES
Aunque la acción más conocida y probablemente la que explica la mayoría de la eficacia de estos fármacos es la que ejercen inhibiendo la activación de los linfocitos T, también actúan sobre los mastocitos, basófilos, los linfocitos B y las células de Langerhans.
En los linfocitos T actúan inhibiendo la acción de la calmodulina, enzima vital en la cadena de activación de estas células que acaba con la producción de interleucina 2 y otras citocinas proinflamatorias.
TACROLIMUS
Estudios sobre eficacia
Son más de 10.000 los pacientes con DA sobre los que se han publicado resultados con tratamiento tópico con tacrolimus.
Los primeros casos de DA tratada con tacrolimus se refieren a pacientes adultos con lesiones recalcitrantes, muy liquenificadas o excoriadas, en cara y cuello. La búsqueda de un inmunosupresor que funcionara de forma tópica y que no presentara los efectos atrofiantes de los corticoides animó a diversos grupos a emplearla en este tipo de pacientes.
Los ensayos abiertos con un corto número de pacientes de Aoyama et al y Nakagawa et al dejaron la puerta abierta al primer ensayo clínico aleatorio, a doble ciego de Ruzicka et al. En éste se comparaba la eficacia y seguridad de las pomadas de tacrolimus al 0,03, 0,1 y 0,3 % frente a placebo. La duración del tratamiento fue de 3 semanas, con 2 semanas adicionales de seguimiento. Se incluyeron 250 pacientes en el estudio con edades entre los 13 y los 60 años. Se aplicaba la pomada en un área predefinida de entre 200 y 1.000 cm2. La mejoría clínica obtenida con cualquiera de los 3 grupos tratados con pomada de tacrolimus al compararla con el grupo que recibió placebo fue mayor de una forma estadísticamente significativa. Sin embargo, no se obtuvo significación estadística al comparar entre sí los tres grupos tratados con tacrolimus, aunque sí una tendencia a favor de los tratados con las concentraciones de 0,3 y 0,1 % frente al 0,03 %. Las lesiones de tronco, miembros, cara o cuello mejoraron de forma similar. El grupo tratado con pomada al 0,03 % redujo en un 66,7 % el marcador global de situación clínica. El tratado con pomada al 0,1 % lo hizo en un 83,4 % y el tratado con pomada al 0,3 % en un 75 %.
El hecho de que los ensayos clínicos previos se habían realizado en adultos, cuando la máxima prevalencia de la DA se da en la infancia, llevó a Boguniewicz et al (18) a iniciar un nuevo ensayo clínico a doble ciego, placebo controlado, en 180 niños con edades comprendidas entre los 7 y los 16 años. De nuevo se compararon los efectos de la aplicación de dos aplicaciones diarias de una pomada de tacrolimus (al 0,03, 0,1 y 0,3 %) y placebo en este caso durante un máximo de 22 días.
Los resultados fueron calificados globalmente como excelentes por los investigadores en un 69, 67 y 70 % de los casos al emplear la pomada de tacrolimus; frente a un 38 % de los casos que usaron placebo (diferencias estadísticamente significativas entre todos los grupos). La media del índice que mide la gravedad de la DA disminuyó de forma dependiente de la dosis en los grupos tratados con tacrolimus (un 72, 77 y 80 %) frente a sólo un 26 % en el grupo de placebo (p < 0,001). Algo similar ocurrió con el prurito.
Paller et al publicaron los resultados de otro ensayo clínico a doble ciego (ECDC) realizado en 351 niños de 2 a 15 años durante 12 semanas. Los niños fueron aleatoriamente tratados con placebo, pomada de tacrolimus al 0,03 % o pomada de tacrolimus al 0,1 %. Se demostraron diferencias estadísticamente significativas en el número de pacientes que alcanzaron una mejoría de al menos el 90 % en la valoración global del médico a favor tanto de la pomada de tacrolimus al 0,03 % como al 0,1 % frente a placebo. Diferencias igualmente significativas se observaron en todos los criterios clínicos objetivos y subjetivos estudiados, así como en la mejoría de la calidad de vida (siempre a favor de las pomadas de tacrolimus). No hubo diferencias significativas en la eficacia demostrada por ambas concentraciones de producto.
Un estudio prácticamente idéntico, pero en 632 adultos ha sido publicado por Hanifin et al. Los resultados de nuevo fueron favorables a las pomadas de tacrolimus pero la de 0,1 % se mostró superior a la de 0,03 %.
Berkesky et al (19) revisan toda la información publicada hasta ese momento en relación a estudios con tacrolimus y DA. En ella recogen igualmente la información vertida en la difícilmente accesible literatura en japonés. De ella se desprende que el porcentaje de pacientes adultos que al menos obtuvieron una mejoría moderada fue mayor en el caso del tacrolimus al 0,1 % (41/41) que con valerato de betametasona al 0,12 % (29/40) (20). En otro estudio similar los resultados fueron igualmente superiores, aunque sin diferencias estadísticamente significativas (21). Al compararlo con tratamientos cortos (1 semana) en la cara o el cuello de adultos los resultados fueron igualmente favorables al tacrolimus: 71 de 73 pacientes al menos mostraron una mejoría moderada en el caso de tacrolimus al 0,1 % frente a sólo 49 de 70 casos tratados con dipropionato de alclometasona (22).
Efectos adversos
En animales de experimentación no se ha demostrado capacidad de producir irritación, fototoxicidad o dermatitis de contacto. En cambio, en humanos sí se ha demostrado una pequeña capacidad de producir sensación de ardor, escozor o calor en la zona de aplicación, particularmente si se hace sobre lesiones en estadio de eczema agudo. Esta irritación es transitoria, dura minutos, aparece sobre lesiones inflamatorias y desaparece según mejoran las lesiones (a los 3 o 4 días como promedio). En el estudio japonés de seguimiento a un año apareció en el 79 % de los pacientes tratados con DA.
En este mismo estudio el 20,7 % de los pacientes presentó algún tipo de infección cutánea, principalmente foliculitis (12 %), eczema herpeticum (4,2 %) o herpes simple (3,3 %). Al realizar un ECDC comparativo entre tacrolimus y placebo a 12 semanas: menos del 6 % de los 631 pacientes adultos desarrollaron foliculitis en el grupo tratado con tacrolimus al 0,1 % frente al 0,5 % de los tratados con placebo (p valor = 0,016) (23).
El contenido de tacrolimus en sangre es suficientemente bajo en la mayoría de pacientes para que el riesgo de efectos adversos sistémicos sea muy bajo. En el 80 % de los casos de pacientes que recibieron tratamiento durante 12 semanas los valores fueron indetectables. Este bajo contenido en sangre hace muy improbable la aparición de efectos adversos sistémicos.
El tacrolimus tópico, a diferencia de los corticoides, no produce atrofia cutánea. In vitro, se demuestra que no afecta la proliferación de los queratinocitos (24, 25), y que no interfiere con la síntesis de colágeno (26). En un estudio publicado por Reitamo et al (27) el tacrolimus, a diferencia del valerato de betametasona, no produce reducción del espesor de la piel tras su uso en humanos durante 7 días cuando se le compara con placebo. Igualmente no disminuye los propéptidos del procolágeno I y III (a diferencia de los corticoides tópicos, que si lo hacen).
El uso prolongado de inmunosupresores por vía parenteral se asocia con un aumento del riesgo de neoplasias, tanto generales como cutáneas. Por lo tanto, existe un posibilidad teórica de aumento del riesgo de presentación de cáncer cutáneo en pacientes que empleen de forma prolongada inhibidores de la función de los linfocitos T. Teóricamente, esto podría ser potenciado por la fotoexposición. Por el momento este hipotético efecto adverso no se ha demostrado en humanos (28).
SDZ ASM 981 (pimecrolimus)
La ascomicina es una de las 23 sustancias macrolactámicas identificadas de los productos de fermentación producidos por el hongo Streptomyces hygroscopicus var. ascomyceticus. A su vez es el compuesto del que se han desarrollado una serie de derivados semisintéticos con una alta capacidad de inhibición de la función de los linfocitos T y la capacidad de ser empleados en forma tópica. SDZ 281-240, ABT-281 y SDZ-ASM981 (pimecrolimus) destacan entre los más prometedores y para los que se han iniciado el desarrollo clínico.
El pimecrolimus inhibe la activación de los linfocitos T mediante la unión a la misma molécula que el tacrolimus, la ciclosporina y el sirolimus, la inmunofilina-12. Por este mecanismo inhibe la calcineurina de linfocitos T e inhibe la liberación de mediadores inflamatorios desde los mastocitos (29).
Estudios sobre eficacia
Van Leent et al (30) publicaron un ECDC de 34 pacientes adultos en los que se probó derecha /izquierda placebo o ascomicina al 1 %. 16 pacientes recibieron dos aplicaciones al día y 18 pacientes una al día. El ensayo duró 3 semanas y mostró una reducción en el marcador global de gravedad del 71,9 % de las zonas en las que se aplicaban dos veces al día ascomicina, del 37,7 % de las zonas en las que se aplicaba una, frente a sólo una reducción del 6,2 % en la zonas de placebo. Las diferencias mostraban diferencias estadísticamente significativas (p < 0,001).
Luger et al (31) publicaron recientemente otro ECDC en los que se estudiaron 260 pacientes adultos que fueron tratados con 4 concentraciones diferentes de crema de SDZ ASM 981, placebo o valerato de betametasona dos veces al día durante 3 semanas. En el estudio quedó demostrada una clara relación dosis-respuesta. Todas las concentraciones del producto en estudio fueron superiores a placebo, siendo la más efectiva la mayor (1 %). La reducción en el marcador global de actividad de la DA (EASI) fue del 53,3 % a las 3 semanas de tratamiento con ascomicina al 1 % frente a un 88,1 % en el caso del valerato de betametasona al 0,1 %.
Harper et al (32) informaron de un ensayo clínico abierto realizado en 10 niños de entre 1 y 4 años de edad tratados con SDZ ASM 981 al 1 % durante 3 semanas. Ocho de los 10 pacientes experimentaron mejoría de entre el 8 y el 89 % en el EASI (un 12,8 % en promedio). Dos de los pacientes tuvieron de ser retirados del estudio el día 10 por rebrote de sus lesiones.
Efectos adversos
Ninguno de los pacientes del estudio de van Leent et al experimentó efectos adversos incluida la irritación cutánea. Sólo 1 de los 8 pacientes que completó el estudio de Harper et al experimentó prurito moderado y transitorio en la zona de aplicación.
En el estudio más amplio de Luger et al el pimecrolimus fue bien tolerado. Los efectos secundarios sistémicos fueron poco importantes y de similar frecuencia en todos los grupos de tratamiento (placebo, pimecrolimus y betametasona). La sensación de ardor, picor, escozor o calor en las zonas de aplicación del tratamiento fue el efecto secundario cutáneo más frecuente. Afectó al 34,9 % de los pacientes tratados con placebo, al 48,9 % de los tratados con pimecrolimus al 1 % y al 9,5 % de los tratados con valerato de betametasona. En la mayoría de casos se inició el primer día y se resolvió hacia el tercer o cuarto día. No se informa de ninguna infección cutánea.
La absorción sistémica del producto es baja, siendo la mayoría de determinaciones de concentraciones plasmáticas del producto indetectables (63 %). La concentración más elevada que se detectó fue de 1,8 ng ml1.
No hay publicado ningún estudio sobre los posibles efectos secundarios del uso prolongado del producto.
INHIBIDORES DE LOS LEUCOTRIENOS
El zafirlukast, el montelukast y el zileuton son tres medicaciones que actúan inhibiendo la acción de los leucotrienos. Reducen la quimiotaxis, la permeabilidad vascular y la broncoconstricción de los casos moderados o leves de asma.
Carrucci et al (33) informaron de que 20 mg/12 h de zafirlucast fueron beneficiosos para 4 adultos con DA graves. Zabawski et al (34) aportan otros 3 casos en adultos y 2 en niños. 20 mg/12 horas de esta medicación permitieron la retirada del resto de medicaciones sistémicas de los pacientes. El seguimiento varió entre las 2 y las 8 semanas. No se informa de efectos secundarios.
Los efectos secundarios de estos preparados son infrecuentes. Incluyen cefaleas y faringitis. Las interacciones medicamentosas son posibles ya que son inhibidores del citocromo P-450.
Yanase y David-Bajar realizaron un ensayo a doble ciego cruzado de montelukast y placebo como tratamiento coadyuvante de la DA durante 4 semanas cada uno (35). Trataron a 8 adultos con DA e informan de una mejoría discreta pero estadísticamente significativa durante el periodo de tratamiento con montelukast al compararlo con el de placebo.
Capella et al (36) publican los resultados de un ensayo clínico simple ciego en el que se comparaba la acción de 10 mg/día de montelukast frente a un régimen combinado (cetirizina oral + claritromicina + corticoides tópicos) durante 6 semanas en 32 pacientes adultos con DA moderada o grave. En este pequeño ensayo no se demostraron diferencias significativas a favor de ninguno de los dos regímenes.
Pei et al demostraron la eficacia del montelukast en la DA de 15 niños de edades entre los 6 y los 16 años con un ensayo clínico a doble ciego frente a placebo (37).
Del tercero de los inhibidores de los receptores de los leucotrienos actualmente comercializados, el zileuton, sólo hemos localizado un trabajo en DA (38). En él se informa de la reducción de los síntomas subjetivos y el eritema de 6 pacientes adultos tratados con zileuton 600 mg/día durante 6 semanas en un ensayo clínico abierto.
