


INTRODUCTION
The evidence for clinical efficacy of sublingual immunotherapy (SLIT) with pollen and house dust mite extracts has been reached in many placebo-controlled, double-blind studies. On the basis of four studies matching strict selection criteria (1-4), the World Health Organisation (5) has recognised the efficacy of this form of therapy but several other DBPC studies with house dust mites (6-8), grass (9, 10), olive pollen (11), Parietaria judaica (12, 13), Artemisia (14), and cat (15) have been published.
The importance of allergy to pollen of trees belonging to the order Fagales (in our case alder, birch, hazel and hornbeam pollen) has been increasing in our region in the last ten years. The prevalence of sensitised patients is currently around 35-40 % among pollic subjects, whose respiratory symptoms are particularly severe from January to May.
In the abundant literature on Specific Immunotherapy, studies on hyposensitisation with extracts of pollen of Betulaceae/Corylaceae (birch and hazel) are uncommon and only a few have been run with non-injective therapies (16-20). Birch pollen has been the first allergen administered by the oral route with enteric-coated capsules, in a study demonstrating the clinical efficacy of this kind of therapy in children (16). Afterwards, only in recent years, one double-blind placebo-controlled study with Betula alba extract administered by sublingual route, has been published (20).
Our study investigated the efficacy and tolerability of SLIT with a mix of Betulaceae/Corylaceae (alder, birch and hazel) pollen extract, administered according to a rush pre-seasonal schedule, followed by a co-seasonal maintenance in patients suffering from rhino-conjunctivitis with or without asthma.
MATERIAL AND METHODS
Study design
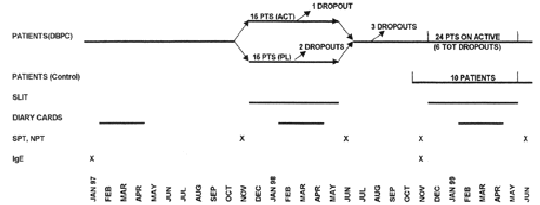
The study followed a double-blind, placebo-controlled design during the first year and an open-controlled design during the second year (fig. 1).
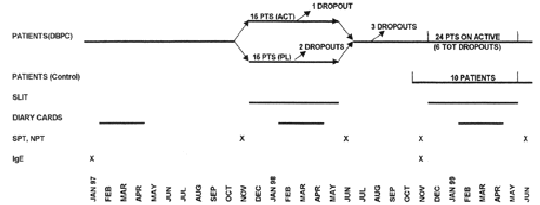
Figure 1.--Study design.
Thirty patients were selected and observed for one pollen season (1997) before immunotherapy to assess the severity of the disease. They were afterwards randomised to receive placebo (15 patients) or active treatment (15 patients) from December 1997 to May 1998 during the first year of the study. Ten patients belonging to the active DBPC Group and 14 belonging to the Placebo DBPC Group gave their availability to be treated with active therapy from December 1998 to May 1999 during the second year of the study. A new group of ten patients, selected at the beginning of 1999 according to the same criteria and treated only with drugs (antihistamine tablets, nasal, bronchial and oral steroids, beta-2 agonists), was considered as a control group during the second year.
Patients' selection
A total of 40 patients were enrolled according to the following inclusion criteria: clinical history of at least two years of seasonal rhino-conjunctivitis, with or without intermittent to moderate persistent asthma (21); sensitisation to Betulaceae/Corylaceae pollens confirmed by in vivo (SPT) and in vitro (RAST) positive diagnosis; age ranging from 12 to 65 years.
Patients were excluded if one of the following conditions recurred: chronic asthma; nasal polyps; previous treatment with specific immunotherapy within the last five years; sensitisation to other inhalant allergens; pregnancy in progress or already planned; chronic or recurrent inflammation of the oral mucosa; other contraindications (22). Sixteen patients suffering from Oral Allergy Syndrome (OAS) associated with the ingestion of some foods were not excluded.
Each patient was informed about aims and procedures of the trial and gave his/her written consent to take part. The experimental protocol was approved by the local Ethics Committee and was notified to the Italian Health Authorities.
In vivo and in vitro diagnosis
At recruitment patients were submitted to SPT with a mixture 1:1:1 of three biologically standardised pollen extracts (alder, birch, hazel), 100 BU/mL of potency, corresponding to an expected wheal size of 75 mm2 on average in sensitised patients (23). A wheal size equal to or larger than the wheal obtained with histamine 10 mg/mL was judged as positive.
Serum specific IgE level was determined according to the RAST EIA technique (Pharmacia, Uppsala, Sweden). Values corresponding to at least class 2 were considered as positive.
Evaluation parameters
Symptom scores
Patients recruited for the DBPC phase filled in a diary for the daily evaluation of symptoms during the period February-April 1997 (before SLIT) and February-April 1998 and 1999 (during SLIT). Patients recruited as control for the open phase were asked to fill in a diary during the period February-April 1999 as well. The scores were reported separately for each of the following five rhino-conjunctivitis symptoms: itching, sneezing, nasal discharge, nasal obstruction, eye itching/redness according to a four grade scale (0 = none, 1 = slight, 2 = moderate, 3 = severe). Asthmatic symptoms were reported according to a 5 grade scale (0 = no symptoms; 1 = cough episodes, no dyspnoea; 2 = repeated cough episodes during day and night, mild dyspnoea; 3 = persistent cough, dyspnoea, nighttime asthma; 4 = persistent cough, asthma during day and night). The maximum daily symptom score was therefore 15 for rhinoconjunctivitis and 4 for asthma.
Drugs
Patients were instructed and allowed to use, if necessary, the following symptomatic medications according to the type and the severity of the disease: antihistamine tablets (loratadine 10 mg/tablet), nasal steroids (beclomethasone dipropionate 50 mg/puff), bronchial steroids (budesonide 400 mg/puff), beta-2 bronchial agonists (salbutamol 100 mg/puff), oral steroids (methylprednisolone 5 mg/tablet).
Each intake of drug for symptom treatment had to be registered daily in the diary card. A specific score was given to each drug: 1 point for each local drug administration (nasal or bronchial steroid, beta-2 bronchial agonist), 2 points for each antihistamine tablet intake and 3 points for each oral steroid tablet intake.
Immunological parameters
Before the beginning of the Sublingual Immunotherapy (SLIT) administration (1997), and at the end of the first year of the study (1998), blood samples were obtained from each patient for specific IgE determination.
Cutaneous reactivity
Before each course of SLIT treatment (November 1997 and 1998) and after the end of each pollen season (June 1998 and 1999), skin reactivity was assessed by SPT in duplicate on the volar surface of the forearms using the same mix of pollen extract (alder, birch and hazel) used for therapy, at three different concentrations: 4, 20 and 100 BU/mL. The wheals were outlined 15 minutes after and transferred onto a paper sheet by means of adhesive tape to measure their size. The Parallel Line Assay (PLA) (24) was used to assess changes in skin sensitivity.
Nasal provocation test (NPT)
The NPT was performed before and after each of the two pollen seasons 1998-1999 (November and June, respectively), using the same extract used for SPT at concentrations 0.5, 1, 2, 4, 8, 16 and 32 BU/mL. After a check with saline solution as negative control, an amount of 80 mL of the 0.5 BU/mL allergen solution was sprayed into each nostril by a metered device. If negative, the test was continued spraying 15 minutes after the following concentration until a positive result was obtained. The reaction was graded according to a 0-3 scale for each of the following symptoms: sneezing, rhinorrea, obstruction and itching (0 = none; 1 = mild; 2 = moderate; 3 = severe). The test was defined as positive when at least two nasal symptoms reached a score of 2.
Pollen count
Pollen count was performed by a 7-day recording volumetric pollen-trap (VPPS 2000, Lanzoni, Bologna, Italy) placed on the flat roof of our hospital (30 m above ground) throughout the study period, according to the Italian Aerobiology Association standardised methodology.
Sublingual immunotherapy (SLIT)
The active specific immunotherapy consisted of a mixture 1:1:1 of three biologically standardised pollen extracts (alder, birch, hazel), graded in six strengths: 0.04, 0.2, 1, 5, 25 and 100 BU/mL in glycerosaline solution. For the birch extract, 100 BU/mL correspond to 66 mg/mL of the major allergen Bet v 1 (25).
Placebo was prepared as saline solution in vials with exactly the same appearance, colour and taste, but without allergens, in order to guarantee the double-blind design of the trial.
All vials contained 50 % glycerine (V/V) and 0,3 % phenol (W/V).
Treatment schedule
A rush pre-seasonal treatment schedule was followed. Drops were taken sublingually twice a day, morning and evening for 18 days starting from 1 drop of strength 0.04 BU/mL up to 5 drops of the same vial. The same procedure was repeated to reach the top dose of 5 drops of strength 100 BU/mL. Each allergen dose had to be kept in the mouth for at least two minutes and then anything remaining in the mouth had to be swallowed (sublingual-swallow technique).
The treatment started in the middle of December 1997 so that the maximum dose was reached before the beginning of the pollen season and repeated 5 days a week (Monday to Friday) for one month. A co-seasonal maintenance treatment followed, at a dosage of 5 drops of strength 25 BU/mL 3 times a week until the end of the pollen season. The cumulative dosage received by each patients was therefore 819 BU in 5 months on average. For birch extract, this amount corresponds to around 445 mg of the major allergen Bet v 1 (26).
Patients were carefully instructed during the first visit in performing the administration and then checked once a month until the end of the trial.
Statistical analysis
Symptoms, drugs intake and NPT data have been statistically analysed by means of non-parametric tests. The Wilcoxon sum rank test has been used for intra-group analysis and the Mann-Whitney U-test for inter-group analysis (BMDP for Windows v. 1. 0, BMDP Statistical Software Inc., Los Angeles, USA.)
Tree-specific IgE were statistically analysed by means of parametric tests (ANOVA).
A specific software was used for the analysis of the skin test reactivity (AIASA, CRS-PLA) (24).
RESULTS
Demographic data
Demographic data of patients enrolled in the trial are shown in table I.
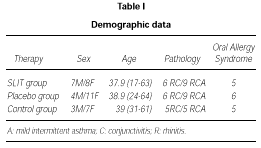
The active and the placebo group of patients for the DBPC phase were well balanced for age, sex, incidence of rhinitis, conjunctivitis and asthma, and OAS.
Twenty-nine patients completed the first year of study, fourteen in the placebo, fifteen in the active group. Only one patient, belonging to the placebo group, dropped out before the end of the DBPC phase because of side-effects (dyspnoea). At the end of the first year, evaluable drug and symptom scores were available for only 27 patients (14 active and 13 placebo), whereas NPT and IgE were available for 29 patients. Twenty-two out of 24 patients submitted to active SLIT during the second year completed the immunotherapy course. One patient dropped-out for exacerbation of rhinitis and OAS during SLIT and another one for personal reasons.
Pollen counts
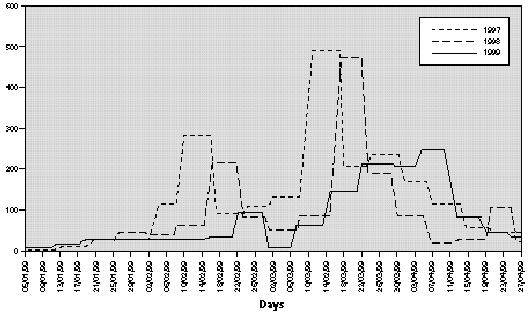
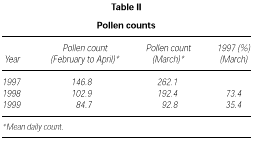
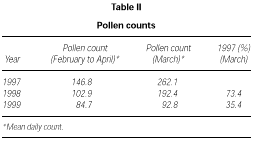
The profile of the pollen curve for Betulaceae/Corylaceae from February to April during the three years of the study was different (fig. 2). The average daily pollen count was higher in 1997 than in 1998 whereas the minimum value was reached in 1999 (table II) both for the full season and for the peak month considered for the evaluation of the symptom and drug scores (March).
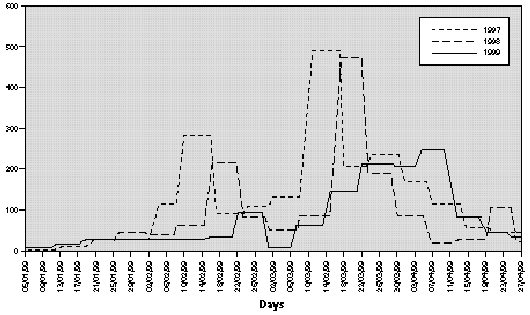
Figure 2.--Grains/m3 tree pollen in the air of Genoa Winter-Spring (1997-1998-1999).
Efficacy parameters
Clinical parameters
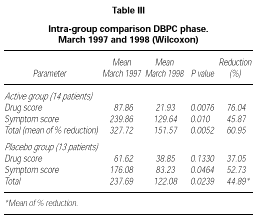
Patients belonging to active or placebo group had well balanced symptom scores (p = 0.139) and drug scores (p = 0.437) at baseline (table III).
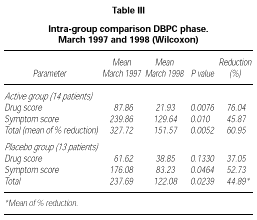
Patients actively treated during the DBPC phase showed during 1998 pollen season statistically significant lower symptom score (p = 0.01), drug score (p = 0.0076) and symptom + drug score (p = 0.0052) compared to the pre-treatment season. No significant difference could be shown in the placebo group for drug score (p = 0.133) whereas a small but still significant difference in this group turned out for symptom score (p = 0.0464) and drug + symptom score (p = 0.0239). No significant differences were found in the inter-group comparison during the DBPC phase. The reduction in drug consumption in the active group was 76.04 % in comparison to a reduction of 37.05 % in the placebo group, whereas the symptom scores were very similar in both groups (table III).
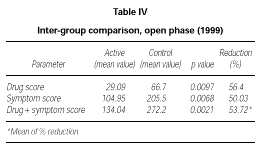
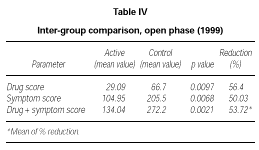
At the end of the second year of SLIT (active treatment for all of the 24 patients) no significant difference was found between patients belonging to the active or placebo group in the previous season (data not shown). Only the subgroup of 10 patients who had received active treatment for two years showed a significant decrease of symptom score (p = 0.0039) and drug consumption (p = 0.0039) and symptom + drug score (p = 0.0166) in comparison to the 1997 season (data not shown). However, from a statistical point of view, on comparing the scores of the actively treated group (22 patients) to the scores of the control group (10 patients) in 1999, a significant reduction could be shown in the first one, not only in symptoms (p = 0,0068) but also in drug intake (p = 0,0097) and symptom + drug (p = 0.0021). The reduction of each parameter was over 50 % (table IV).
Objective parameters
Skin reactivity (PLA) did not show a statistically significant change within each group before (1997) and at the end of the first year of therapy (1998) (data not shown).
Only in patients who received the active treatment, an increase in the tolerated dose in nasal challenge (NPT) was reported at the end of the study, but the difference was not statistically significant (data not shown).
Specific IgE level did not show any statistically significant variation (data not shown).
Side effects
Eleven patients (7 belonging to the active group, and 4 to the placebo group) reported mild local side effects (itching of lips and oral mucosa) during the DBPC phase. One patient under placebo dropped out because of dyspnoea whereas one patient reported exacerbation of food-related OAS and rhinitis after a short active SLIT period (dropped-out during the second year). No other general side effect of any importance was reported in any group during the whole trial.
DISCUSSION
We chose to use a mix of alder, birch and hazel pollen extract because of the large cross-reactivity in the Betulaceae/Corylaceae family (27, 28).
Tree pollen allergy has been studied until now in Northern European countries, where birch pollen is the most represented, in spite of a very short period of diffusion. In other countries the exposure to other pollen species of the family (alder, hazel and hornbeam) can provoke symptoms lasting some months and interrupted by asymptomatic periods. This could make the analysis of the clinical efficacy of the therapy more difficult than in patients sensitised to allergens with a long and/or continuous period of exposure such as mites, Parietaria and grass, partially explaining the rarity of studies on this issue.
Only one DBPC study with sublingual therapy for birch has been so far published (20), but a comparison with our study is difficult because of the somewhat artificial environment (Vienna Challenge Chamber) patients were exposed to for assessment.
In our study, the efficacy of a sublingual-RUSH immunotherapy with a standardised tree-pollen extract was assessed in comparison to placebo in the first phase and to a control group of adult patients suffering from rhinoconjunctivitis, with or without asthma, due to sensitisation to these pollens. According to the EAACI Position Paper about the optimal study design for Immunotherapy (29), we included a pre-treatment monitoring of symptoms and drug score for one season. In fact, pre-selection of patients for our study was done during the immediately preceding pollen season, when they started the filling-in of diary-cards.
According to the selection criteria used for the DBPC phase, all patients (30/30) suffered from rhinoconjunctivitis and only some of them (18/30) suffered also from mild to moderate/persistent asthma. For this reason, the scoring system adopted was mainly focussed on rhinoconjunctivitis symptoms (maximum daily score 15) instead than asthma (maximum daily score 4). Because this leads also to a higher total score for symptoms as compared to score for drugs, the total reduction in table III was calculated as a mean of the percentage of reduction of each parameter instead of as a mean reduction on the total symptoms + drug scores.
Compared to the pre-treatment pollen season (1997), the first year of study showed a significant decrease in drug-score, symptom-score and symptoms + drug score in the actively treated group. A lower but still significant decrease in symptom score and symptoms + drug score, but not in the drug score, was obtained in the placebo group. This quite strong placebo effect, as pointed out by Malling (30), combined with the low statistical power of the analysis due to the reduced number of patients observed (27 in total) and also to a lower pollen load during the DBPC season as compared to the pre-treatment observation, led to a non significant difference in the inter-group (active-placebo) comparison.
In our opinion this interpretation is supported by the analysis of the second part (open-controlled) of the study where the placebo effect should have played a minor role. During the second year of treatment we observed statistically significant lower overall scores in the active group (22 patients) in comparison to the control group (10 patients).
The analysis of the intra-group evolution showed a further, significant decrease in symptom-scores for patients already submitted to the active therapy during the first year, but this result was obtained with a lower pollen count in comparison to the previous two years. This result seems to be indicative of the need or opportunity of at least two or more years of active treatment to get better results, as already stated for injective immunotherapy.
Among the other evaluation parameters considered, skin sensitivity did not show any significant variation before and after SLIT. In other studies (4, 11) a decrease in skin reactivity was recorded only after a longer period of treatment without interruptions (18-24 months), while in our trial patients received a pre- and co-seasonal SLIT course for a short period (4-5 months/year). The difference in treatment schedules is in our opinion a good explanation for the different results.
The same reason could explain why, in spite of a significant reduction of the nasal symptom score and an increase in the tolerated dose of allergen in NPT, a statistically significant intra-group difference could not be demonstrated. In a similar study with Parietaria pollen (5), we have found a significant decrease in nasal reactivity, but patients in that trial had been treated without interruption for 24 months.
During the study, one patient belonging to the placebo group and one to the active group developed OAS, whereas patients already suffering before the trial from this syndrome showed no change.
The side effects noted in this study were negligible, as found in most studies including mainly adult patients and in one study with children (31). No severe systemic reactions were observed in any group whereas itching of lips and oral mucosa was reported after administration of active therapy or placebo to a similar extent. These outcomes are obviously in favor of both the safety of SLIT and the adequate blinding of the study.
We may conclude that SLIT with tree pollen extract is a safe treatment, by a rush schedule too. The assessment of the clinical efficacy seems to be more difficult than in other studies. The evaluation of objective data (skin-reactivity, NPT) could have been influenced by the short duration of therapy, whereas the evaluation of subjective results appears to have been clearly influenced by the placebo effect, particularly relevant in this kind of studies. The inclusion of a control group not receiving placebo can be useful for a better analysis of the outcomes.
