


The innate immune system possesses a network of germline–encoded receptors that recognize microbial molecular motifs and endogenous molecules produced by injured tissues and set in motion a defensive response which adapts to the damage that has occurred. This network includes Toll-like receptors (TLRs), a family of transmembrane receptors that recognize a wide spectrum of ligands at the cell surface and in the lumen of intracellular vesicles. Recognition of ligands by TLRs induces the recruitment of different cytoplasmic adaptor molecules and initiates signalling pathways which ultimately lead to the activation of transcriptional factors such as NF-κB, IRF1/3/5/7, or AP-1. These factors are involved in the expression of inflammatory cytokines, chemokines, type I interferons, co-stimulatory molecules, and other factors of the effector response. TLRs regulate many aspects of both innate and adaptive immunity. To prevent an inappropriate or an overactive immune response, a complex network of molecules negatively regulates TLRs and their associated signalling pathways. TLRs are currently viewed as important targets for the development of new vaccines and innovative therapies which may help prevent or treat disorders such as cancer, allergy, autoimmunity, obesity, atherosclerosis, and other inflammatory diseases.
Most living species have developed efficient mechanisms of surveillance and defence that have protected them from the pathogens they face. These defensive mechanisms are mediated by products encoded in the host genome and constitute a phylogenetically preserved immunity, known as innate immunity1. The cells of innate immunity possess a set of germline-encoded receptors (termed pattern recognition receptors or PRRs), and each receptor harbours a fixed specificity that enables it to detect invariant molecular constituents of the pathogens (known as pathogen-associated molecular patterns, or PAMPs) and initiate a defensive response which is adapted to the pathogen2,3. Besides their primary function in discriminating infectious non-self from non-infectious self, PRRs can also identify endogenous molecules released during tissue injury, and it has been postulated that, rather than sense non-self, PRRs detect “danger signals” and alert the immune system4. Different experimental models propose that some of these innate sensors monitor specific physiological disturbances, and PRRs are believed to be involved in the control of cell and tissue homeostasis5–7.
The strategy used by innate immunity to mount a particular effector response is based on the existence of a set of different PRRs in all cells of the same type that can discriminate between ligands. Functional cooperation and complementation exist between these receptors, thus providing a combinatorial repertoire of signals that triggers and shapes the immune response best suited to destroying a specific pathogen and repairing the damage produced8,9.
To date, three families of PRRs that activate signalling pathways and induce the expression of inflammatory genes have been identified10: Toll-like receptors (TLRs), nucleotide binding oligomerisation domain (NOD)-like receptors (NLRs), and retinoic acid inducible gene-I (RIG)-like receptors (RLRs). TLRs were the first to be described and are the most widely studied. In this review, we examine the main structural and functional features of TLRs, their ligands, and the signalling pathways that they activate. We also highlight the importance of TLR cooperation for the induction of a specific immune response.
DISCOVERY OF THE TLR FAMILYTLRs are a family of innate receptors that have been evolutionarily conserved from plants to mammals. They derive their name from the Drosophila Toll protein, with which they share a sequence and functional similarity. This protein was initially involved in the embryonic development of Drosophila11,12; however, given the remarkable similarity between the signalling pathway of Toll protein and mammalian LRs, the interleukin (IL)-1 receptor signalling pathway which leads to the activation of NF-κB (Fig. 1), it was proposed that Toll protein might be involved in the fly's immune defence against pathogens13,14. Several subsequent experimental works in mutant flies clearly showed that Toll protein was involved in protecting the fly against infection by fungi and gram-positive bacteria15,16. These studies revealed the existence in invertebrates of a highly conserved defensive strategy able to discriminate between various classes of microorganisms in order to mount a response adapted to the pathogen, despite the fact that invertebrates only have innate immunity. When these results were published, a human homologue of Toll protein (later named TLR4) was shown to regulate the expression of NF-κB-controlled inflammatory genes and the expression on antigen-presenting cells of the co-stimulatory molecules required for the activation of naive T cells. These results showed that TLRs not only mediate in the effector response of innate immunity, but they also participate in the functional polarization of naive lymphocytes during antigen cross-presentation17. TLR4 was later identified as a lipopolysaccharide (LPS) sensor18,19. Subsequently, a growing family of evolutionarily conserved proteins that were structurally related to Drosophila Toll and could recognize a broad spectrum of PAMPs and activate immunity were identified.
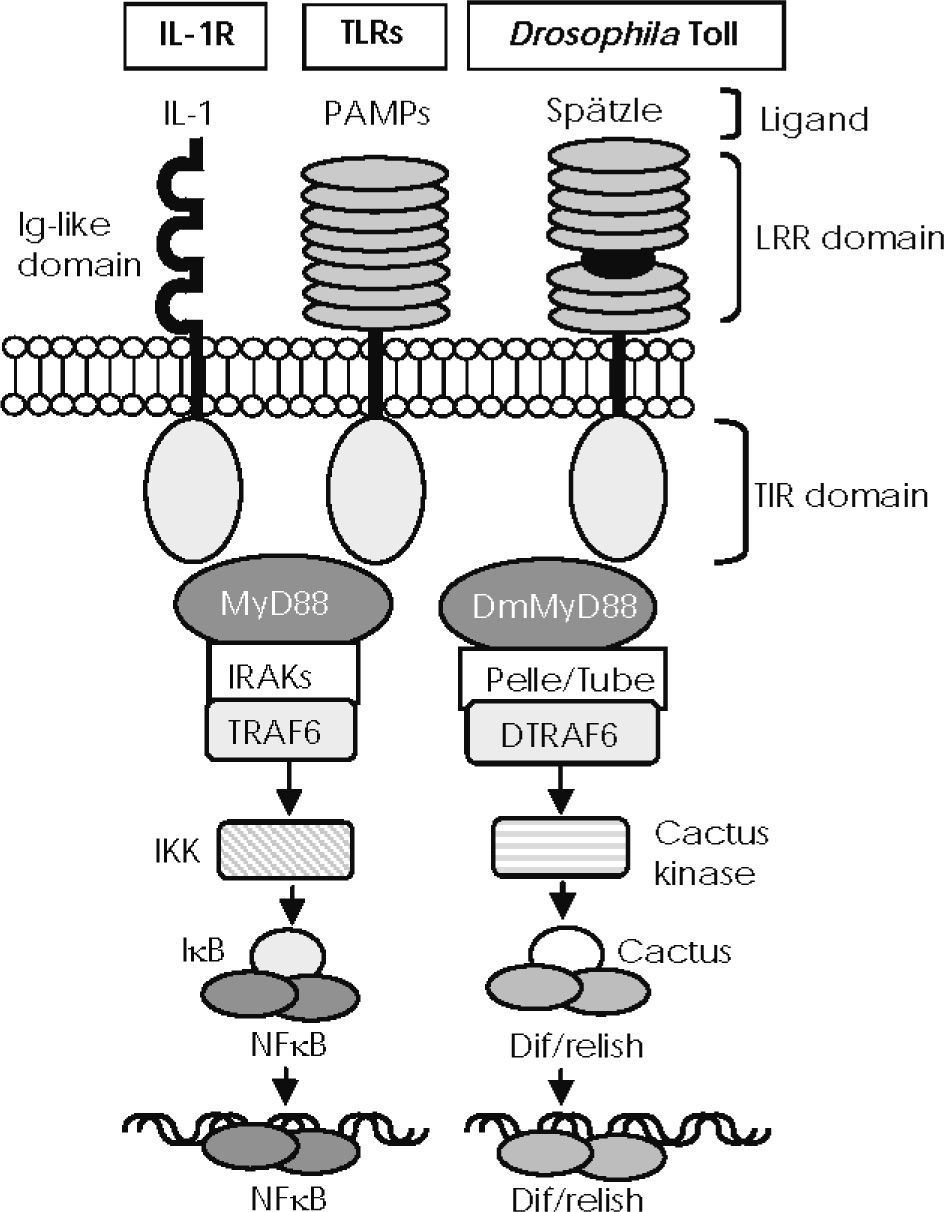
 and Drosophila Toll possess an extracellular domain containing different leucine-rich repeat (LRR) motifs, which detect pathogen-associated molecular patterns (PAMPs) and the Spätzle molecule, respectively. IL-1 receptors recognise IL-1 members through an immunoglobulin-like structure. They all share a cytoplasmic signalling domain called the Toll/IL-1 receptor domain (TIR). TIRs of mammalian TLRs and of IL-1 receptors associate with MyD88 and recruit members of the interleukin-1 receptor–associated kinase (IRAK) family, leading to TRAF6 activation to degrade IκB and activate NF-κB. Activated Drosophila Toll associates with DmMyD88 (a homologue of mammalian MyD88) through the TIR domain to recruit signalling molecules such as Tube and Pelle (a serine-threonine kinase that is highly homologous to mammalian IRAKs) and activate DTRAF6 (homologue of TRAF6 in the fly), thus inducing the degradation of Cactus (the Drosophila IκB homologue) and the release of Dif/relish dimer, a transcription factor homologue of NF-κB.")
—IL-1R/Toll-like receptor superfamily structure and conserved signalling pathways. Mammalian Toll-like receptors (TLRs) and Drosophila Toll possess an extracellular domain containing different leucine-rich repeat (LRR) motifs, which detect pathogen-associated molecular patterns (PAMPs) and the Spätzle molecule, respectively. IL-1 receptors recognise IL-1 members through an immunoglobulin-like structure. They all share a cytoplasmic signalling domain called the Toll/IL-1 receptor domain (TIR). TIRs of mammalian TLRs and of IL-1 receptors associate with MyD88 and recruit members of the interleukin-1 receptor–associated kinase (IRAK) family, leading to TRAF6 activation to degrade IκB and activate NF-κB. Activated Drosophila Toll associates with DmMyD88 (a homologue of mammalian MyD88) through the TIR domain to recruit signalling molecules such as Tube and Pelle (a serine-threonine kinase that is highly homologous to mammalian IRAKs) and activate DTRAF6 (homologue of TRAF6 in the fly), thus inducing the degradation of Cactus (the Drosophila IκB homologue) and the release of Dif/relish dimer, a transcription factor homologue of NF-κB.
To date, 13 different TLRs have been characterized in mice and 11 TLR homologues (10 of which are functional) have been found in the human gene database20,21. Based on their amino acid sequence and function, human TLRs are subdivided into five subfamilies: TLR2, TLR3, TLR4, TLR5, and TLR922. The TLR2 subfamily has four members: TLR1, TLR2, TLR6, and TLR10, which are highly homologous and work in a pair-wise combination in the presence of their respective ligands. The TLR9 subfamily is composed of TLR7, TLR8, and TLR9. The subfamilies TLR3, TLR4, and TLR5 include only one member that can work alone or in association with other receptors or molecules.
Most tissues express mRNA for at least one TLR and several express all of them23, with considerable differences between mice and humans24. In both species, the most abundant repertoires of TLR are detected in the cells and tissues involved in innate immunity and in those tissues highly exposed to the environment. At the cellular level, TLRs are differentially distributed and their location correlates with the nature of their ligands25,26. Members of the TLR2, TLR4, and TLR5 subfamilies are mainly located in the plasma membrane and recognize external ligands. The TLR3 and TLR9 subfamilies are located in the membranes of intracellular vesicles such as endosomes, and only detect ligands present in the lumen of these intracellular structures. This distribution is not permanent, and the expression and location of TLRs can vary depending on cell and tissue status27–29.
During the last ten years, several experimental works have revealed that TLRs are key regulators of both the innate response and the adaptive response. The TLRs on the sentinel cells of innate immunity initiate and regulate inflammation30, on dendritic cells they play an essential role in the polarization of the adaptive immune response31, and on the cells of adaptive immunity they regulate their functions32.
TLR STRUCTURE AND SIGNALLING PATHWAYSMost of the proteins that participate in cellular signalling networks are constructed in a cassette-like fashion and contain several protein interaction domains. Some of these domains have the intrinsic ability to undergo homotypic or heterotypic domain-domain interactions to identify a physiological partner involved in a common signalling process and form functional complexes33.
TLRs are integral membrane glycoproteins that activate signalling pathways through interaction domains. Structurally, TLRs are characterized by the presence of an ectodomain involved in ligand recognition, which consists of leucine-rich repeat (LRR) motifs and a cytoplasmic signalling domain (Toll/IL-1 receptor [TIR] domain) that shows a remarkable similarity to that of the IL-1 receptor family, which is essential for signal transduction. Both domains are joined by a single transmembrane helix22.
In Drosophila, TLRs do not interact directly with PAMPs, but are activated upon binding a cleaved form of the cytokine-like Spatzle molecule. During the immune response, this protein is thought to be processed by serine proteases secreted in hemolymph that are activated by the recognition of gram-positive bacteria or fungi34. Binding of Spatzle to TLRs induces recruitment of a cytoplasmic adaptor, initiating a cascade of recruitments and phosphorylations analogous to the mammalian NF-κB activation pathway (Fig. 1).
In mammals, the structural basis of ligand recognition is still poorly understood, and there is a relative lack of experimental evidence to support a physical interaction between LRR domains and TLR ligands. In any case, upon detection of PAMPs, TLRs dimerise and/or associate with other receptors or molecules and induce the interaction between the TIR-cytoplasmic domain and the TIR- domains of intracellular adaptor molecules35. This in turn initiates a cascade of recruitments that results in the successive activation of different members of the IL-1 receptor-associated kinase (IRAK) family. These events ultimately lead to the activation of transcriptional factors such as NF-κB, interferon (IFN) regulatory factor (IRF)-1/3/5/7, and/or activator protein-1 (AP-1), which allow the expression of genes that code for inflammatory cytokines, chemokines, type I IFNs, co-stimulatory molecules, and other factors of the effector response36.
Four intracytoplasmic TIR-containing adaptors involved in TLR signalling have been identified: myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor inducing IFN-β (or TRIF, also named TICAM1), TIR domain-containing adaptor protein (or TIRAP, a structurally related MyD88 protein also known as MyD88-adaptor-like (MAL) protein), and TRIF-related adaptor molecule (or TRAM, also known as TICAM2)21,36. MyD88 and TRIF mediate in the activation of two independent signalling pathways that can be categorised as the MyD88-dependent pathway and the TRIF-dependent pathway. TIRAP and TRAM act as “bridging molecules” with TIRA protein recruiting MyD88 to TLR2 and TLR4, and TRAM recruiting TRIF to TLR4.
MyD88-dependent signalling pathwayMyD88 is the key signalling adaptor for all TLRs except TLR3, but it is also the adaptor molecule for the IL-1 receptor family (see Figs. 1 and 2). MyD88 has a bipartite nature, containing an N-terminal death domain and a COOH-terminal TIR domain. Upon stimulation with appropriate ligands, the TIR domain of the cytoplasmic portion of the TLRs directly associates with the TIR domain in the C-terminal portion of MyD88. In the case of TLR2 and TRL4, TIR-dependent recruitment of MyD88 requires TIRAP to facilitate TIR domain interactions37. The death domain in the N-terminal region of MyD88 is involved in the recruitment and successive phosphorylations of different members of the IRAK family21,35. IRAK4 and IRAK1 are sequentially phosphorylated and then dissociated from MyD88 to interact with TNF-associated factor 6 (TRAF6), which in turn activates transforming growth factor (TGF)-β-activated protein kinase 1 (TAK1), a protein that, in combination with TAB1, TAB2, and TAB3 proteins, mediates in two different downstream pathways. One of these pathways is involved in the activation of AP-1 transcription factor through the mitogen-activated protein (MAP) kinase cascade. The other pathway activates the IκB-kinase (IKK) complex, which is composed of two catalytic subunits, IKKa and IKKp and of a regulatory subunit IKK7 (also known as NF-κB essential modulator or NEMO), which catalyses the phosphorylation and degradation of IκB, thus enabling activation of NF-κB. Active AP-1 and NF-κB transcription factors then translocate to the nucleus and mediate in the expression of genes that regulate inflammatory cytokines such as TNFα, IL-6, IL-1 β, or IL-12, and several chemokines21,35,36. The MyD88-IRAK-TRAF6 complex directly phosphorylates IRF-5 to be translocated to the nucleus and induces gene expression of proinflammatory cytokines, such as IL-6, IL-12, and TNFα38. Stimulation of TLR9 by their specific ligand also induces the recruitment of TRAF3 to the complex formed by MyD88, IRAK-4, IRAK-1, and TRAF6, to phosphorylate and activate IRF7, the main inducer of type I IFNs39. MyD88 also binds to IRF1, when it is induced by IFNγ, and travels assembled to the nucleus to induce expression of IFNβ and IL-12 p3540.
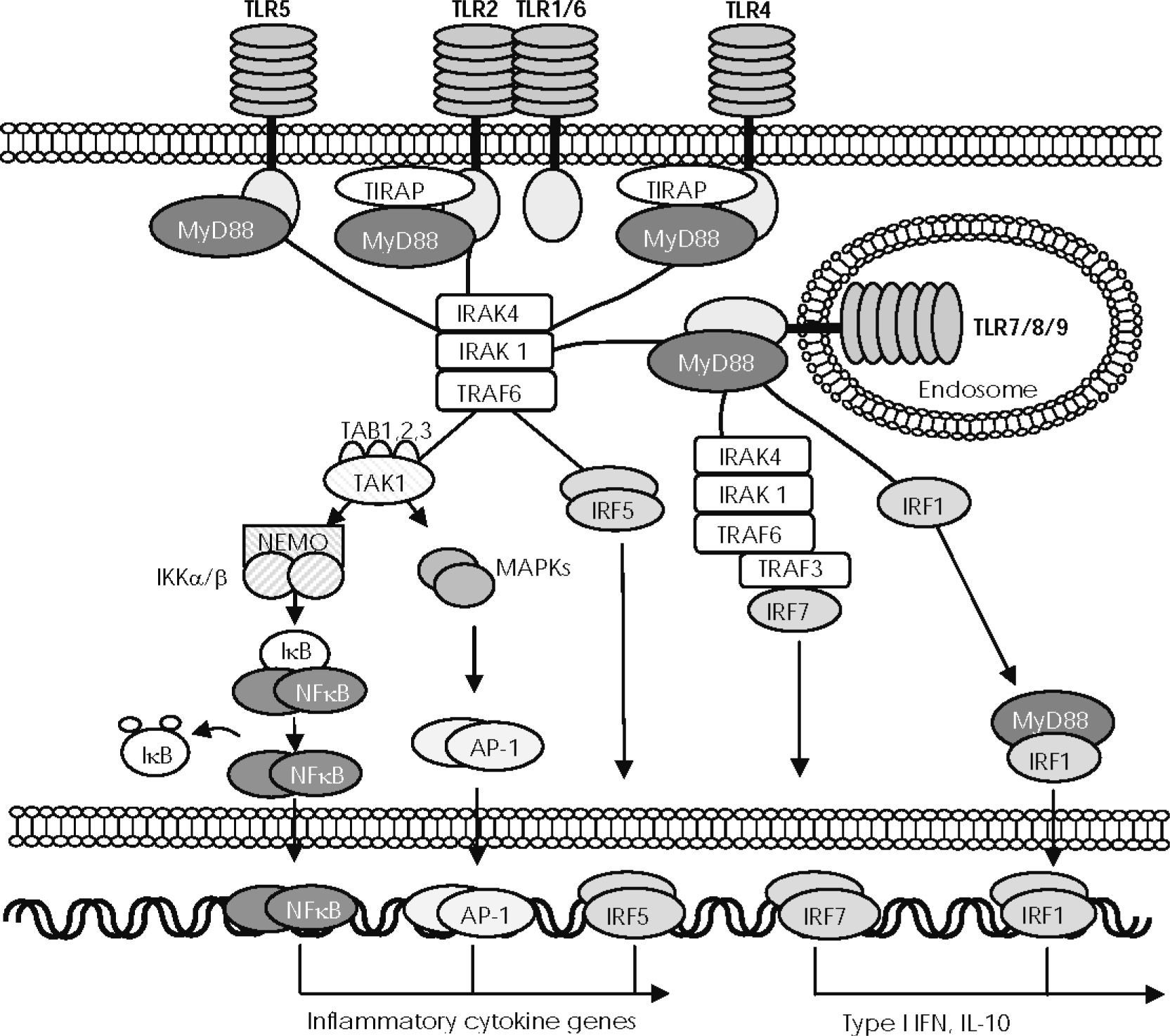
, except TLR3, induces the recruitment of myeloid differentiation factor 88 (MyD88) through association with the Toll/IL-1 receptor (TIR) domain, although TLR2 and TLR4 require TIR domain–containing adaptor protein (TIRAP). MyD88 then recruits different members of the interleukin-receptor–associated kinase (IRAK), which interacts with TNF-associated factor 6 (TRAF6) to activate transforming growth factor-β–activated protein kinase1 (TAK1). Next, TAK1 combines with TAB1, TAB2, and TAB3 proteins to mediate in two different pathways of downstream signals. One pathway activates the IκB-kinase (IKK) complex (composed of IKKα, IKKβ, and regulatory subunit IKKγ/NEMO, which allows NF-κB to translocate to the nucleus. The other pathway activates the MAPK pathway, which mediates AP-1 activation and translocation. IFN regulatory factor (IRF) 5 can be recruited to the MyD88-IRAK4-TRAF6 complex to be phosphorylated and directly translocated to the nucleus. NF-κB, AP-1, and IRF5 control the expression of genes encoding inflammatory cytokines. In plasmacytoid dendritic cells (pDC), stimulation of TLR9 induces the recruitment of TRAF3 to the complex formed by MyD88, IRAK-4, IRAK-1, and TRAF6, to phosphorylate and activate IRF7, the main inducer of type I IFNs. MyD88 also combines with IRF1 when it is induced by IFNγ and travels assembled to the nucleus to induce the expression of IFNβ.")
—The MyD88/TIRAP-dependent signalling pathway. Activation of toll-like receptors (TLRs), except TLR3, induces the recruitment of myeloid differentiation factor 88 (MyD88) through association with the Toll/IL-1 receptor (TIR) domain, although TLR2 and TLR4 require TIR domain–containing adaptor protein (TIRAP). MyD88 then recruits different members of the interleukin-receptor–associated kinase (IRAK), which interacts with TNF-associated factor 6 (TRAF6) to activate transforming growth factor-β–activated protein kinase1 (TAK1). Next, TAK1 combines with TAB1, TAB2, and TAB3 proteins to mediate in two different pathways of downstream signals. One pathway activates the IκB-kinase (IKK) complex (composed of IKKα, IKKβ, and regulatory subunit IKKγ/NEMO, which allows NF-κB to translocate to the nucleus. The other pathway activates the MAPK pathway, which mediates AP-1 activation and translocation. IFN regulatory factor (IRF) 5 can be recruited to the MyD88-IRAK4-TRAF6 complex to be phosphorylated and directly translocated to the nucleus. NF-κB, AP-1, and IRF5 control the expression of genes encoding inflammatory cytokines. In plasmacytoid dendritic cells (pDC), stimulation of TLR9 induces the recruitment of TRAF3 to the complex formed by MyD88, IRAK-4, IRAK-1, and TRAF6, to phosphorylate and activate IRF7, the main inducer of type I IFNs. MyD88 also combines with IRF1 when it is induced by IFNγ and travels assembled to the nucleus to induce the expression of IFNβ.
The adaptor molecule TRIF is used by TLR3 and TLR4 to initiate an MyD88-independent signalling pathway that leads to activation of NF-κB, IRF-3, and IRF-721,35,36, but that is also an inducer of apoptosis41 (Fig. 3). Ligand binding to TLR3 and TLR4 results in TRIF recruitment through its TIR domain, although TLR4 requires cooperation with TRAM to bind to TRIF42. The N-terminal region of TRIF possesses a domain that recruits two non-canonical IKKs—IKKi and TANK-binding kinase (TBK) 1—to phosphorylate IRF3 and IRF7, which form homodimers and translocate from the cytoplasm to the nucleus, where they regulate expression of high levels of IFNβ and of different host defence genes. It is thought that TRIF associates with TBK1 through TRAF343. Recently, it has been proposed that IRF3 activated by the TRIF-dependent pathway promotes the synthesis of TNFα, which then binds to its receptor to initiate a late phase of NF-κB activation in an autocrine manner44,45.
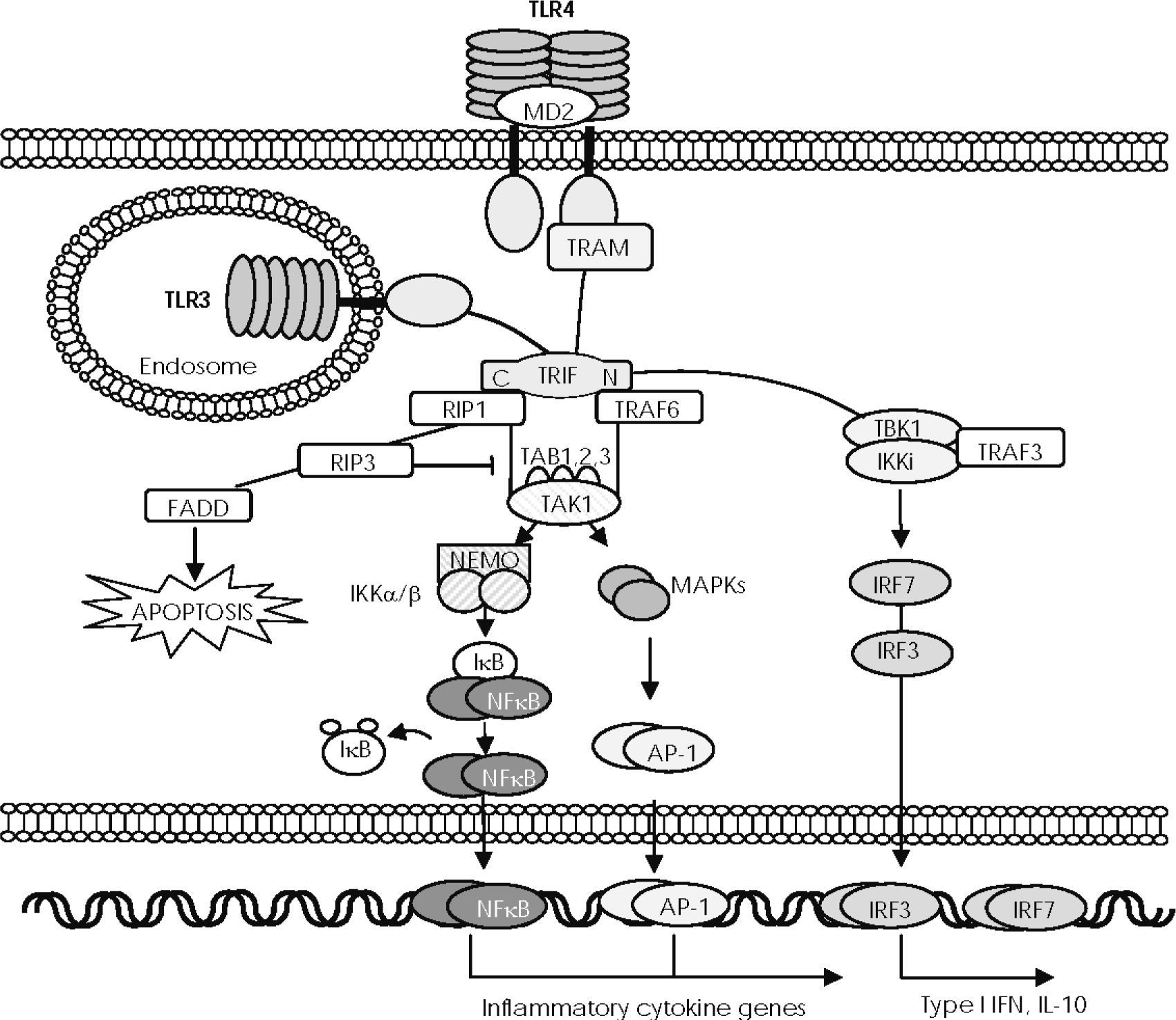
 3 and TLR4 recruit Toll IL-1 receptor (TIR) domain–containing, adaptor-inducing interferon (IFN)-beta (TRIF) through the TIR domain, although TLR4 requires the TRIF-related adaptor molecule (TRAM). The N-terminal region of TRIF directly recruits the non-canonical IκB kinases (IKKs), TBK1 and IKKi, which mediate phosphorylation and dimerisation of IRF3 to be translocated to the nucleus and induce expression of IL-10 and type I IFN genes. The N-terminal region of TRIF also recruits TRAF6 and the C-terminal region mediates its interaction with receptor interacting protein (RIP) 1. RIP1 then forms a complex with TRAF6 and TAK1, resulting in activation of NF-κB and MAPKs. RIP3 blocks RIP1-induced NF-κB activation and the death domain of RIP1 recruits Fas/Apo-1–associated death domain protein (FADD), thus inducing apoptosis.")
—The TRIF-dependent signalling pathway. After exposure to their respective ligands, Toll-like receptor (TLR) 3 and TLR4 recruit Toll IL-1 receptor (TIR) domain–containing, adaptor-inducing interferon (IFN)-beta (TRIF) through the TIR domain, although TLR4 requires the TRIF-related adaptor molecule (TRAM). The N-terminal region of TRIF directly recruits the non-canonical IκB kinases (IKKs), TBK1 and IKKi, which mediate phosphorylation and dimerisation of IRF3 to be translocated to the nucleus and induce expression of IL-10 and type I IFN genes. The N-terminal region of TRIF also recruits TRAF6 and the C-terminal region mediates its interaction with receptor interacting protein (RIP) 1. RIP1 then forms a complex with TRAF6 and TAK1, resulting in activation of NF-κB and MAPKs. RIP3 blocks RIP1-induced NF-κB activation and the death domain of RIP1 recruits Fas/Apo-1–associated death domain protein (FADD), thus inducing apoptosis.
The N-terminal region of TRIF contains another domain that mediates interaction with TRAF6. The C-terminal region of TRIF interacts with the receptor interacting protein (RIP) 1 and forms a complex with TRAF6 and TAK1. Together, the recruitment by TRIF of RIP1 and TRAF6 seems to facilitate TAK1 activation, which in turn results in activation of NF-κB and of MAP kinases. When RIP3 is complexed with TRIF, RIP1 blocks RIP1-induced NF-κB activation and the death domain of RIP1 recruits Fas/Apo-1-associated death domain protein (FADD) to induce apoptosis45.
NEGATIVE REGULATION OF TLR SIGNALLINGAlthough TLR-mediated signalling pathways are indispensable for the eradication of microbes and tissue repair, their prolonged and/or excessive activation would lead to an aberrant immune response and disease. To avoid or minimise inappropriate reactivity or an excessive immune response, TLR signalling is tightly regulated through multiple negative regulatory mechanisms, which seem to be non-redundant46,47 (Fig. 4). The first line of regulation is to avoid improper or excessive interaction between TLRs and their ligands. Excessive binding of ligands can be prevented by naturally produced soluble TLRs, which function as decoy receptors48,49, by reducing TLR expression or by inducing their degradation50,51. Other molecules directly hamper binding to the receptor52. Once the TLR and ligand have interacted, TLR signalling can be further controlled by intracellular regulators which could be upregulated by TLR signalling during damage or constitutively expressed to physiologically control inflammation. These molecules can inhibit TLR signalling pathways at different levels (Fig. 4).
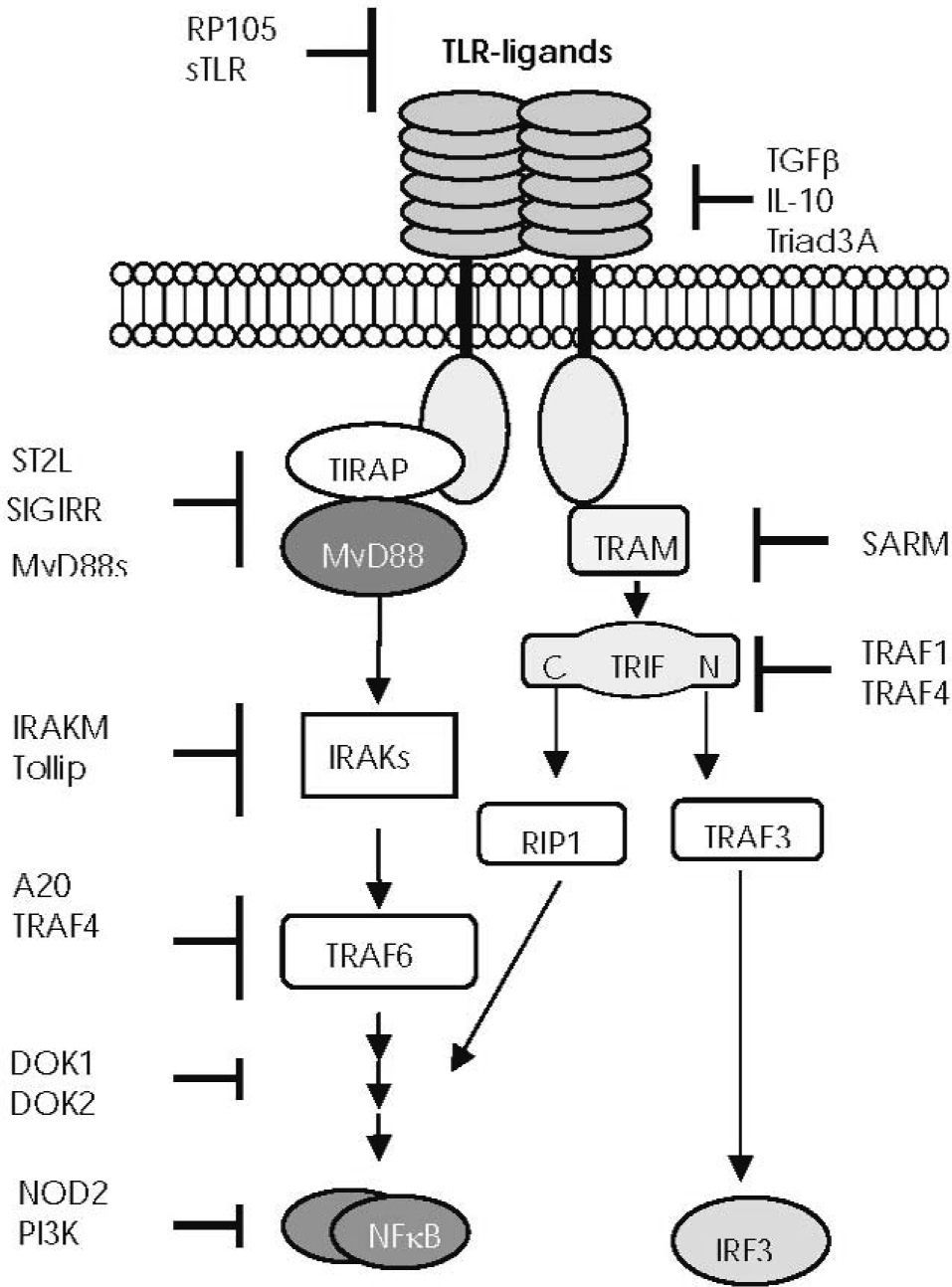
 function. Endogenous molecules interfere in the TLR downstream signalling pathways at different levels: soluble TLRs (sTLR) and the radioprotective 105 protein (RP105) prevent excessive binding of ligands to TLR52. Transforming growth factor beta (TGF-β), IL-10, and Triad 3A regulate TLR expression. The short form of MyD88 (MyD88s) antagonises MyD88 function53. The membrane-bound form of the IL-1 receptor-like protein (known as ST2L) sequesters MyD88 and TIRAP, thus preventing their association with the TIR domains54. The single immunoglobulin IL-1 receptor- related molecule (SIGIRR, also known as TIR8) interacts with the TIR domain of the TLR455. IL-1 receptor–associated kinase M (IRAKM) seems to prevent the activation of the tumour necrosis factor receptor–associated factor 6 (TRAF6)56. Toll-interacting protein (TOLLIP) interacts with IRAK157. The zinc finger protein A20 acts by blocking TRAF6 signal58. The inhibitory proteins downstream of kinase 1 and 2 (DOK1 and DOK2) negatively regulate Ras-Erk signalling downstream of protein tyrosine kinases59. TNF receptor–associated factor (TRAF) 1 interacts with the TIR domain of TRIF and inhibits TLR3-mediated activation of NF-κB60, whereas TRAF4 acts as a silencer in TLR-mediated signalling through its association with TRAF6 and TRIF61. The sterile-alpha and HEAT/armadillo motif–containing protein (or SARM), seems to associate with TRIF62. PI3K (phosphatidylinositol 3-kinase) is a negative regulator of TLR signalling, but its molecular basis is still unclear63–65. Balanced production of pro-inflammatory cytokines mediated by the TLR2 signalling pathway seems to be regulated by NOD2 through an L-10-mediated anti-inflammatory cytokine response66.")
—Main points of negative regulation of Toll-like receptor (TLR) function. Endogenous molecules interfere in the TLR downstream signalling pathways at different levels: soluble TLRs (sTLR) and the radioprotective 105 protein (RP105) prevent excessive binding of ligands to TLR52. Transforming growth factor beta (TGF-β), IL-10, and Triad 3A regulate TLR expression. The short form of MyD88 (MyD88s) antagonises MyD88 function53. The membrane-bound form of the IL-1 receptor-like protein (known as ST2L) sequesters MyD88 and TIRAP, thus preventing their association with the TIR domains54. The single immunoglobulin IL-1 receptor- related molecule (SIGIRR, also known as TIR8) interacts with the TIR domain of the TLR455. IL-1 receptor–associated kinase M (IRAKM) seems to prevent the activation of the tumour necrosis factor receptor–associated factor 6 (TRAF6)56. Toll-interacting protein (TOLLIP) interacts with IRAK157. The zinc finger protein A20 acts by blocking TRAF6 signal58. The inhibitory proteins downstream of kinase 1 and 2 (DOK1 and DOK2) negatively regulate Ras-Erk signalling downstream of protein tyrosine kinases59. TNF receptor–associated factor (TRAF) 1 interacts with the TIR domain of TRIF and inhibits TLR3-mediated activation of NF-κB60, whereas TRAF4 acts as a silencer in TLR-mediated signalling through its association with TRAF6 and TRIF61. The sterile-alpha and HEAT/armadillo motif–containing protein (or SARM), seems to associate with TRIF62. PI3K (phosphatidylinositol 3-kinase) is a negative regulator of TLR signalling, but its molecular basis is still unclear63–65. Balanced production of pro-inflammatory cytokines mediated by the TLR2 signalling pathway seems to be regulated by NOD2 through an L-10-mediated anti-inflammatory cytokine response66.
TLRs can also function as death receptors and induce apoptosis, a process that might be important in the control of a deregulated TLR response67. This novel mechanism of activation-induced cell death could also play an important part in the resolution of inflammation.
TOLL-LIKE RECEPTOR LIGANDSRelatively few TLRs have been identified to date; however, they enable the innate immune system to recognise an increasingly broad spectrum of ligands from bacteria, fungi, protozoa, viruses, and many endogenous molecules.
Cell surface TLRs mainly recognise microbial membrane lipids68. The TLR2 subfamily has been shown to identify lipoproteins and lipopeptides from a wide range of pathogens including gram-negative bacteria, mycoplasma and spirochetes69–72, peptidoglycans and lipoteichoic acid from gram-positive bacteria27,73,74, lipoarabinomannan from mycobacteria75, and LPS from non-enterobacteria76,77. TLR4 mainly recognizes LPS, but can also sense mannans from Saccharomyces cerevisiae and Candida albicans78, glucuronoxylomannan from Cryptococcus neoformans79, and taxol80, and it can mediate in the response to respiratory syncytial virus81. Mammalian TLR5 recognises bacterial flagellin, the principal component of bacterial flagella from both gram-positive and gram-negative bacteria82. Intracellular TLRs mainly sense nucleic acids and their derivatives. Human TLR3 recognises foreign-derived double-stranded RNA (dsRNA) and participates in the generation of protective immunity against some viral infections83. Natural agonists consisting of nucleic acids, such as single-stranded RNA (ssRNA) or DNA with unmethylated CpG motifs, activate innate immune cells through TLR7/8 or TLR9, respectively84. TLR9 also recognises haemozoin, a pigment from the malaria parasite Plasmodium, and seems to play an important role in the cerebral pathology of malaria85.
Other evidence shows that non-microbial molecules can be detected by TLRs and activate an inflammatory response. TLR2 and TLR4 recognise the release of degradation products by damaged tissues as danger signals86. These endogenous ligands include molecules such as heat shock proteins, fibronectins, hyaluronic acid, heparan sulfate, fibrinogen, defensin, surfactant protein A, minimally modified low-density lipoprotein (LDL), and extracellular matrix degradation products. In addition, several studies clearly reveal that TLR7 and TLR9 are sensors of damage and enhance the immune response by sensing endosomally translocated self-DNA or self-RNA87,88, and human TLR3 recognises endogenous necrotic cell RNA. It has been proposed that, in certain autoimmune disorders, recognition of endogenous ligands by TLRs drives sterile inflammation sustained by innate immune cells that contributes to the loss of tolerance89. Similarly, it is interesting to emphasize that many autoantigens are generated by tissue injury and are able to stimulate innate immunity through TLRs. This supports the idea that many of them are autoantigens because they act as autoadjuvants that directly activate innate immunity to induce a self-directed immune response87.
Recent reports show that TLR2 and TLR4 recognise saturated fatty acids and activate both MyD88-dependent and TRIF-dependent pathways to induce the expression of pro-inflammatory gene products, whereas various unsaturated fatty acids inhibit TLR-mediated signalling pathways and target gene expression90. It has been proposed that TLRs sense pathological levels of lipids playing a role in the central control of energy homeostasis, and it is speculated that the detection of abnormal levels of dietary lipids through TLRs could participate in the control of food intake7. These new concepts in innate immune function can help us to understand the pathophysiological basis of conditions such as obesity, insulin resistance, or atherosclerosis90.
SPECIFIC RECOGNITION OF LIGANDS BY TLRSIn spite of the low number of TLRs identified, their role in the identification of invading pathogens and of injury produced is essential for the induction of a specific protective response. One explanation for the capacity of such a reduced number of TLRs to discriminate between a large number of stimuli is that these receptors assemble into heteromeric or homomeric complexes and/or associate with other receptors or with other co-molecules to form many different activation clusters91,92. These combined associations enable the discrimination of ligands and modulate TLR signalling properties. For example, heterodimerisation of TLR2 with TLR1 or TLR6 permits the innate immune system to differentiate between the numerous lipopeptide structures present in different pathogens93. Furthermore, CD14, a membrane receptor expressed mainly on phagocytes, is required for TLR2/6 signalling to both lipopeptides and zymosan94,95. The scavenger receptor CD36, a membrane receptor which binds fatty acids and directs their transfer into the cells, contributes to the identification of TLR2/6 ligands containing diacylglycerides, thus facilitating their signalling96. Another example of TLR molecular cooperation is the recognition of LPS by TLR486,92. The LPS binding protein (LBP) binds to the amphipathic lipid A moiety of LPS, thus facilitating its presentation and transfer to CD14, which guides the complex to TLR4. Next, the myeloid differentiation 2 protein (MD-2) acts as an extracellular adaptor that binds to the hydrophobic portion of LPS and to the extracellular domain of TLR4. This MD-2 interaction determines molecular rearrangements in TLR4 and their homotypic aggregation, and triggers transmission of signals. When endotoxin is transferred from MD-2 to an extracellular soluble CD14, it attenuates TLR4-dependent cell activation by acting as a negative regulator.
FUNCTIONAL COOPERATION OF TLRSPathogens contain many different PAMPs that interact with a specific combination of TLRs expressed by the different host cells. Simultaneous activation of TLRs can generate cross-talk between receptors and modify the primary responses of a TLR to their agonist, thus adding a further level of complexity to the regulation of innate immunity. Simultaneous activation of signalling pathways will result in complementary, synergistic, or antagonistic effects97. For example, synergy has been described between MyD88-associated and TRIF-associated pathways for the induction of several proinflammatory cytokines. Antagonistic cooperation has been seen in human dendritic cells: the subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in these cells is blocked by IL-10 released by concomitant TLR2 stimulation. Other forms of TLR cooperation seem to promote negative regulation of their signalling pathways.
A TLR-mediated response can also interact with responses induced by ligands of other PRR families and cooperate in the induction of the host defensive response97. Moreover, many types of cells participate in the response to injury, and PRR stimulation activates a myriad of genes, leading to the release of mediators that can exert potent autocrine and paracrine effects. Indeed, different authors have shown that the same TLR ligand can have different effects on the host cell under the influence of different signals mediated by accessory cells and other immunocytes97,98.
At present, it is unknown how the ligands of TLRs that activate signalling pathways which converge on activation of common transcription factors, such as NF-κB or AP-1, are coordinated to regulate specific gene expression and generate a pathogen-specific response. Recently, a greater understanding of these subtleties has become evident with interesting new insights into specific regulation of different NF-κB subunits at the transcriptional and functional levels99. Other authors have found evidence that a post-translational modification of NF-κB and the cross-talk of NF-κB with other cooperating transcription factors play a role in the fine regulation of a specific transcriptional response at the promoter level100.
FURTHER PERSPECTIVESThe identification of TLR ligands and their signalling pathways and regulatory mechanisms has opened an array of possibilities in the development of innovative vaccines and therapies to prevent and treat infection, cancer, allergy, and a long list of immunological disorders101,102, many of which are currently being evaluated in clinical trials103–106. However, the immune system is different to other drug targets and its modulation could trigger unexpected harmful responses107. Accordingly, although TLRbased therapies have an enormous biological potential, their benefits are not free of risk107–111. In order to prevent the undesirable adverse effects of these therapies, we still need a more precise understanding of the participation of TLRs in the pathophysiology of a disease before drugs enter the trial phase and routine clinical practice. We still need to expand our knowledge on TLR cross-talk with other receptors, the influence of cells from the microenvironment, and the way in which the signals generated are coordinated to elicit a specific response. We also need to determine how many endogenous ligands signal through TLRs and ascertain their role in human health. In conclusion, the success of these potent biological therapies will require the efforts of multidisciplinary teams including immunologists with detailed knowledge of potential side effects.
I am grateful to Thomas O'Boyle for editorial advice.
