



Leucocyte adhesion deficiencies (LADs) are a group of primary immunodeficiencies in which the leucocytes are unable to migrate from the circulation towards the areas of inflammation. Three types of LAD have been described to date1–3:
- 1.
Type I leucocyte adhesion deficiency (LAD I), characterised by mutations in the common chain (CD18) of the β2 integrins family. These patients suffer serious recurrent infections of the skin and mucosal membranes. In the more serious presentations the patients die early if haematopoietic precursor cell transplantation is not carried out.1–3
- 2.
Type II leucocyte adhesion deficiency (LAD II), characterised by the absence of the fucosylated ligand in neutrophils needed for binding to selectins E and P in the activated endothelium. Clinically, these patients suffer less serious infections but present retarded psychomotor and weight and body height development.1–3
- 3.
Type III leucocyte adhesion deficiency (LAD III), characterised by a defect in the activation of integrins β1, β2 and β3. These patients suffer serious infections and bleeding disorders.1–3
We present a case of type I leucocyte adhesion deficiency (LAD I).
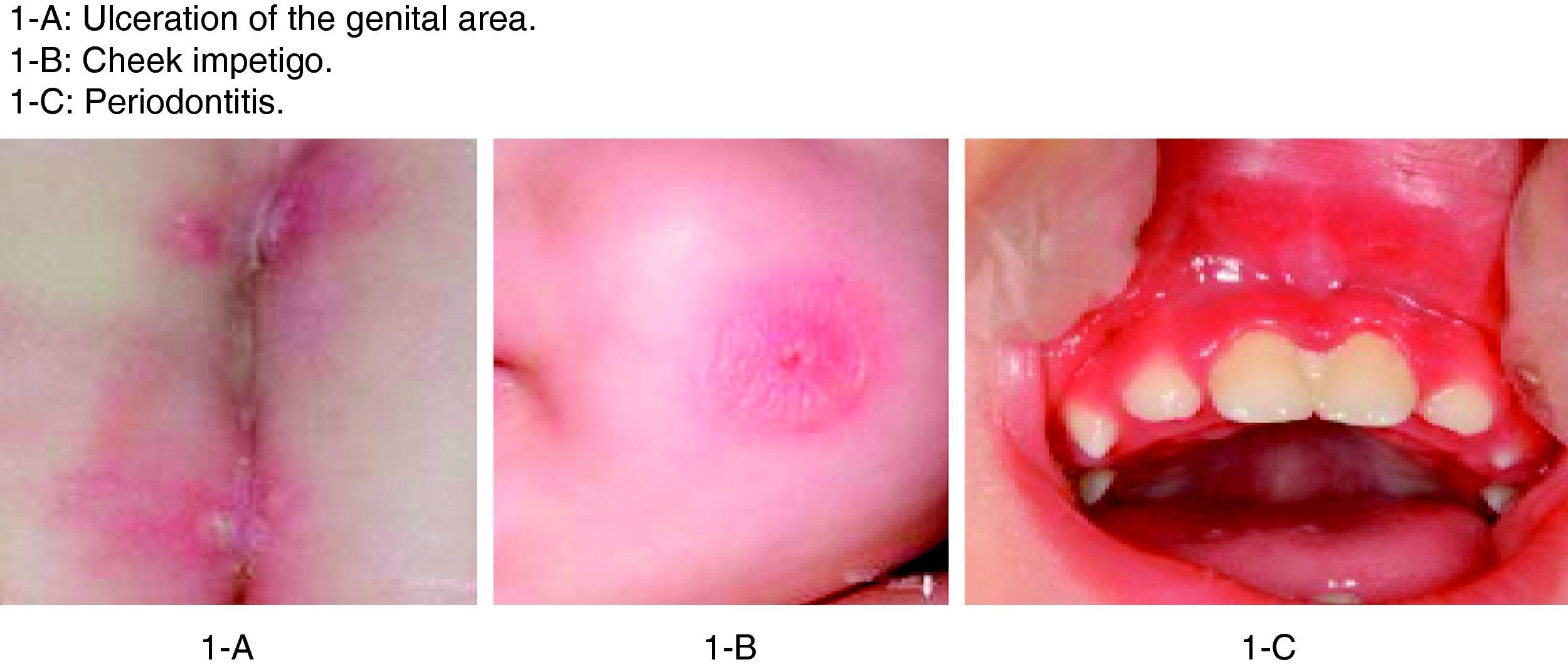
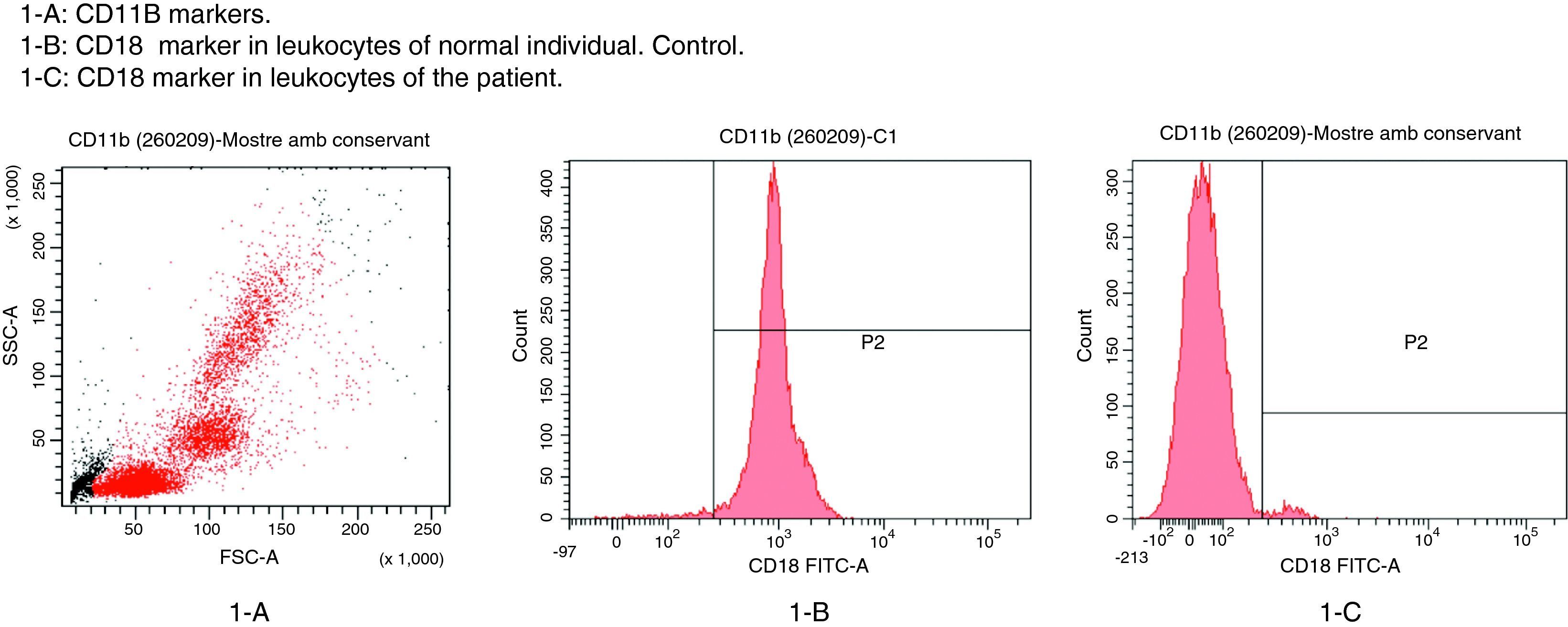
The patient in this case was a 3-month-old boy, the first offspring of consanguineous parents (first cousins). There had been no previous miscarriages. The female first cousin of the parents had died 15 days after birth due to non-established causes. Pregnancy and delivery were without complications. The patient was born to term with a body weight concordant with the gestational age. Weight and height progression was normal. Seven days after birth the patient was admitted due to omphalitis, with culture positive for penicillin-sensitive Streptococcus mitis and multisensitive Escherichia coli. Blood culture proved negative, and the complete blood count showed 42,500leucocytes/mm3 with a normal formula. At 2 months of age the patient was again admitted due to urinary infection caused by multiresistant E. coli and staphylococcal impetigo. At 3 months of age he was admitted due to left-side acute otitis media. The complete blood count showed 33,600leucocytes/mm3 (56.9% neutrophils and 31.6% lymphocytes). Two weeks later the patient developed an ulceration in the lumbar and intergluteal zone that again required admission to hospital. The patient was found to be in good general condition, with a weight of 6kg and no fever. A rounded, ulcerated non-suppurative lesion with an erythematous margin was confirmed in the lumbar and intergluteal zone (Table 1 and Fig. 1). Blood tests: leucocyte count 26,500cells/mm3 (31% neutrophils and 53.9% lymphocytes), C-reactive protein 6mg/l, erythrocyte sedimentation rate 11mm/h, with negative blood and lesion sample cultures. Empirical antibiotic treatment was started with meropenem. An immune study was carried out, revealing the following lymphoid population distribution: LB 18%, LT 62%, LT4 46%, LT8 15%, absolute LT4 6578/mm3. IgM: 3038mg/l, IgG: 4627mg/l, IgA: 437mg/l, IgE: 47kU/l. Neutrophil oxidative capacity test 96%, as determined by flow cytometry with dihydrorhodamine. Leucocyte adhesion deficiency (LAD) was suspected, as a result of which flow cytometry with anti-CD11/CD18 monoclonal antibodies was carried out, revealing the absence of CD18 in leucocytes (Fig. 2). The blood group corresponded to A+ (discarding group hh Bombay present in type II leucocyte adhesion defect). An ITGB2 gene mutation analysis was performed, revealing the presence of genetic mutation p.Gly-169-Arg (also known as p.G169R) in exon 5 of the mentioned gene and in both alleles (homozygosis). Given the compatible clinical manifestations, the total absence of CD18 expression in peripheral blood leucocytes, and the presence of mutation p.G169R, we concluded that the patient suffered a severe type I leucocyte adhesion defect. Study of both parents was decided on in order to establish the segregation pattern of the detected mutation. Flow cytometric analysis of both parents revealed CD18 present in 98% of the leucocytes, while mutation analysis of the ITGB2 gene showed the presence in both parents of the same mutation identified in the patient, though in only one of the two alleles (heterozygosis) (Table 2).
Clinical manifestations of LAD I.
| Clinical manifestations | |||
| Age | Clinical manifestation | Microorganism | Treatment |
| 7 days | Omphalitis | Streptococcus mitisEscherichia coli | Ampicillin–gentamycin |
| 1 month | Fever syndrome | Negative cultures | Amoxicillin–clavulanate |
| 2 months | Urinary infectionImpetigo | Escherichia coliStaphylococcus aureus | Cefuroxime |
| 3 months | Acute otitis media | Amoxicillin–clavulanate | |
| 3 months | Genital ulcer | Negative cultures | Meropenem |
| 4 months | Cheek impetigo | Staphylococcus aureus | Amoxicillin–clavulanate |
| 6 months | Urinary infectionAcute otitis media | Escherichia coli | Ceftriaxone |
| 7 months | Acute otitis media | Amoxicillin–clavulanate | |
| 10 months | Impetigo | Negative cultures | Amoxicillin–clavulanate |
| 17 months | Periodontitis | Negative cultures | Vancomycin–meropenem |


Non-specific (innate) immunity study in LAD I.
| Non-specific immunity study, adhesion molecules | |
| CD18 in leucocytes (flow cytometry) | Absent |
| Blood group | A+ |
| Genetic study | |||||||
| Gene | Disease | Exons studied | Results | Nucleotide substitution | Amino acid change | Status | |
| Patient | ITGB2 | Type I LAD | 1→15 | 2 mutations | c. 505 G→A | p.Gly-169-Arg | Homozygosis |
| Father | ITGB2 | Type I LAD | Exon 5 | 1 mutation | c.505 G→A | p.Gly-169-Arg | Heterozygosis |
| Mother | ITGB2 | Type I LAD | Exon 5 | 1 mutation | c.505 G→A | p.Gly-169-Arg | Heterozygosis |
The patient posteriorly required further admissions due to impetigo on two occasions, urinary infection caused by E. coli, and periodontitis – with persistently elevated leucocyte and neutrophil counts.
The definitive diagnosis was type I leucocyte adhesion deficiency (LAD I), with severe phenotype: mutation p.G169R in exon 5 of the ITGB2 gene in homozygosis. Healthy parents carrying the mutation in heterozygosis.
Early antibiotic therapy was started on occasion of all recurrences, followed by a good clinical course. Regarding aetiological treatment, HLA typing of the patient was started, together with the search for a haematopoietic precursor cell donor. At the age of 19 months peripheral blood allogenic haematopoietic precursor cell transplantation from an unrelated donor was carried out (Id 9/10 molecular). Transplantation was started with non-myeloablative conditioning treatment administering CAMPATH-1 at a dose of 0.2mg/kg/day i.v. for 5 days, fludarabine 30mg/m2/day i.v. for 5 days, melphalan 140mg/m2 i.v. and allopurinol p.o. 300mg/m2/day for 7 days. Prophylaxis against graft-versus-host disease was provided with cyclosporine 1.5mg/kg/12h i.v. from day −1 to day +30, plus mycophenolate 7.5mg/kg/12h from day −1 to day +30. The patient moreover also received prophylactic antibiotic, antiviral and antifungal therapy. Haematopoietic precursor cell infusion was carried out without complications, and followed by haematological recovery. The patient posteriorly developed infectious complications caused by CMV (with PCR positive for CMV), for which gancyclovir was prescribed, as well as adenovirus infection (coproculture positive for adenovirus) for which cidofovir was prescribed, and infection due to Aspergillus (galactomannan antigen positive in blood), for which voriconazole was administered. Haematological toxicity was moreover observed, requiring red cell and platelet transfusions. The patient developed acute cutaneous, gastrointestinal and hepatic graft-versus-host disease, receiving treatment with prednisone, topical triamcinolone, Leukotac and tacrolimus. Death occurred two and a half months after transplantation as a result of these complications.
A non-specific (innate) immune response is triggered as a result of infection. The neutrophils and monocytes are attracted from the peripheral circulation towards the site of infection, binding to adhesion molecules located on the endothelial cells, and responding to chemotactic factors produced as a consequence of the infection.4 This phenomenon takes place in four phases:
- (1)
Extension of the leucocytes over the endothelium, due to the action of the selectins which rapidly increase their expression on the surface of the endothelial cells of the postcapillary veins at the site of infection. The two types of selectins expressed by the endothelial cells are selectin P and E. A third type called selectin L is expressed by lymphocytes and other leucocytes. The selectins allow the neutrophils to adhere to the endothelial cells, which in turn have been activated through the action of cytokines (TNF, IL1 and IFNγ) present in the inflammatory focus.
- (2)
Increased integrin affinity as a result of chemokine activity. The chemokines stimulate cell chemotaxis. The integrins are heterodymer compounds of covalently bonded α and β protein chains.4
- (3)
Leucocyte adhesion to the endothelium. The cytokines also favor endothelial ligand expression, particularly vascular cell adhesion molecule-1 (VCAM-1, a ligand for integrin VLA-4), and intercellular adhesion molecule-1 (ICAM-1, a ligand for integrins LFA-1 and Mac-1). This leads to free binding of the leucocytes to the endothelium.4
- (4)
Transmigration of the leucocytes through the endothelium: The chemokines act upon the adhered leucocytes and stimulate cell migration through the inter-endothelial spaces towards the site of infection.4
An alteration in any of the steps of the above process will result in defective leucocyte migration.
The present clinical case corresponds to a type I leucocyte adhesion defect due to mutation p.G169R in exon 5 of the ITGB2 gene in homozygosis, exhibiting an autosomal recessive hereditary trait.
LAD I is an autosomal recessive disease resulting from a quantitative or qualitative defect in the common chain (CD18) of the β2 integrins family. Over 300 cases have been reported to date.2 The leucocytes of patients with LAD I are deficient in terms of the expression of the three integrins that contain CD18:
- •
Lymphocyte function antigen-1 (LFA-1, CD11a/CD18).
- •
Mac-1 (CD11b/CD18).
- •
Gp 150/95 (CD11c/CD18).
The primary defect in this disease is related to the β2 subunit, the biosynthesis of which is necessary for surface expression of the α subunits.2
The patient showed a total absence of CD18 expression in peripheral blood leucocytes, thus compromising the expression of these three integrins belonging to the β2 integrins family.
In vitro studies have shown important defects in chemotaxis, adhesion and migration through the endothelial cell layer. This genetic disorder is due to mutations in the gene of the β2 integrin (ITGB2) encoding for the CD18 subunit, located at an extremity of the long arm of chromosome 21q22.23. The molecular bases underlying the CD18 deficiencies are variable. There have been descriptions of mutations that lead to the expression of quantitatively normal CD18 but with functional defects – although few such cases have been documented. Point mutations have been described that lead to defective protein synthesis with the substitution of a single amino acid. Other mutations lead to splicing defects, resulting in the production of unstable proteins – many of which are located at the binding site of the β2 integrins to their ICAM 1 or 2 ligands. In turn, other genetic defects result in a reduction in CD18 mRNA expression. Other cases are characterised by the expression of mRNA or proteic precursors of aberrant size, leading to large or small CD18 subunits. An important percentage of the CD18 mutations identified in LAD I are located in the extracellular domain of the CD18 subunit, in exon 9 – this being a highly preserved region. Although LAD I can be caused by a range of mutations, all result in the production of a non-functional β2 subunit. In most cases small point mutations, minor insertions or deletions are reported in the ITGB2 gene.1,2
Our patient presented the mutation p.Gly-169-Arg (glycine substitution by arginine in position 169) in exon 5 of the ITGB2 gene located on the long arm of chromosome 21, previously described in the literature as a mutation responsible for severe forms of type I leucocyte adhesion defect.5,6
LAD I is clinically characterised by delayed shedding of the umbilical stump, recurrent bacterial infections mainly of the skin and mucosal membranes, leucocytosis, periodontitis, the absence of pus and poor wound healing.1,7–9
The severity of the infectious complications in patients with LAD I appears to be directly related to the degree of CD18 deficiency. Two phenotypes have been described that make it possible to classify the condition as corresponding to either severe or moderate deficiency disease1,2:
- •
Severe: less than 2% of the normal expression of CD18. These patients suffer earlier, frequent and serious infections, and most die in childhood if appropriate treatment is not provided.
- •
Mild to moderate: CD18 expression ranges from 2 to 30% of normal. These patients suffer few serious infections, and most survive into adult life.
Our patient presented all the clinical manifestations corresponding to the severe form of the disease.
Patients of this type have difficulties defending themselves against bacteria and fungi, although they do not show an increased susceptibility to viral infections.2 The infections affect the skin, respiratory tract, intestine and perirectal area usually from the time of birth. The lesions can become necrotic, ulcerating and often quickly progress towards systemic infection. A classical infectious presentation of the disorder is omphalitis, with delayed shedding of the umbilical stump (>30 days). Severe gingivitis and periodontitis are the most frequent infections among those patients who survive beyond childhood. The most commonly implicated microorganisms are Staphylococcus aureus and enteric gramnegative bacilli.1–3
Our patient suffered several infectious episodes caused by S. aureus and E. coli, and on one occasion S. mitis was isolated.
The absence of pus at the sites of infection is characteristic of the disease.1 Such patients have a severely limited extravascular leucocyte mobilisation capacity, as a result of which these cells are unable to reach the sites of inflammation. Biopsies of the infection sites reveal inflammation without neutrophils. In turn, there is a diminished presence of lymphocytes in the lymphoid tissue, since LFA-1 plays an important role in the function of these cells. Late shedding of the umbilical stump is one of the expressions of the poor wound healing that characterises these individuals, and is accompanied by a dystrophic appearance of the resulting scar tissue. Laboratory tests can show moderate neutrophilia in the absence of infection. During the infectious process, marked leucocytosis with neutrophilia (5–20 times greater than normal, reaching counts of up to 100,000cells/mm3) is observed, due to the incapacity to mobilise the cells towards extravascular inflammatory sites.2 This was characteristically seen in our patient.
The diagnosis of the disease should be suspected in all infants with recurrent soft tissue infections and important leucocytosis. The diagnostic criteria were established in 199910:
- 1.
Definitive diagnosis: patients with diminished CD18 expression in neutrophils (<5% of normal), and at least one of the following:
- -
Mutation of the β2 integrin gene.
- -
Absence of mRNA encoding for β2 integrin in leucocytes.
- -
- 2.
Probable diagnosis: patients with diminished CD18 expression in neutrophils (<5% of normal), and all of the following:
- -
Recurrent bacterial or fungal infections.
- -
Leucocytosis >25,000cells/mm3.
- -
Delayed shedding of the umbilical stump and/or wound healing defects.
- -
- 3.
Possible diagnosis: patients with important leucocytosis >25,000cells/mm3 and one of the following:
- -
Recurrent bacterial infections.
- -
Deep-seated and severe infection.
- -
Absence of pus at the sites of infection.
- -
Confirmation of the diagnosis requires demonstration of the absence of CD18 and the associated alpha subunit CD11a, CD11b and CD11c on the surface of the leucocytes, based on flow cytometry using monoclonal antibodies CD11 and CD18.1 Sequence analysis is advised in all cases in order to define the molecular defect of the β2 subunit. Leucocytes express CD18 at surface level from week 20 of intrauterine development; as a result, cordocentesis performed after this time can contribute to establish a prenatal diagnosis.2 In those families in which a molecular defect has been identified, an early prenatal diagnosis can be established via chorionic biopsy and mutation analysis. Recently, a pre-implantation diagnosis has been established.11
In most cases the clinical manifestations and laboratory test findings are suggestive of the disease and the diagnosis is clear. However, a differential diagnosis is required with conditions also characterised by important leucocytosis, such as infections, leukaemoid reactions, leucaemia and other lymphoproliferative processes.1
The treatment of the disease depends on the severity of the clinical picture. In the case of the mild to moderate phenotype, the infections respond to conservative management and the early and appropriate use of antibiotics during the acute episodes. Correct oral hygiene is important for the control of periodontitis and for preventing infections. These patients can receive all vaccines, including those involving live viruses. Prophylactic antibiotic treatment can reduce the risk of infection, and adequate management of the infectious processes can allow such patients to survive into adulthood.1,2
In the case of patients with the severe disease phenotype, the only corrective treatment available to date is the transplantation of haematopoietic precursor cells.1,2,12,13 The absence of LFA-1 in these patients can constitute an advantage for transplantation, since graft rejection appears to depend in part on the CD18 complex. The largest series published to date describes 36 children in 14 centres subjected to transplantation between 1993 and 2007, and followed-up on for 5 years after transplantation. The reported survival rate was 75%. Low-intensity conditioning regimens were found to be safe.12 Transplant success depends on early diagnosis and treatment, the conditioning regimen used before transplantation, and the degree of donor compatibility. Gene therapy has been evaluated in the context of preclinical and in vivo trials, inserting a normal ITGB2 gene in haematopoietic stem cells, with promising results.14 However, further studies are needed to support the results obtained. These patients die in childhood if transplantation is not carried out as soon as possible. If transplantation is performed before serious infections develop, the resulting prognosis is very good.
To summarise, our patient presented the typical clinical characteristics of type I leucocyte adhesion deficiency (LAD I), i.e., omphalitis, late shedding of the umbilical stump, recurrent infections of the skin and mucosal membranes without pus formation, persistently elevated leucocyte counts and flow cytometric findings indicative of the severe phenotype of the disease. The diagnosis in turn was confirmed by the genetic study. Although LAD I is a rare form of congenital immune deficiency, it must be taken into account in patients who present this clinical picture.
