








El calcio es el mineral más abundante en el organismo y junto con el fósforo constituye el componente inorgánico mayor del hueso. Para mantener una homeostasis adecuada, se requiere una compleja interacción entre factores hormonales y no hormonales, y el correcto funcionamiento de sus órganos diana, en particular riñón, intestino y hueso, además de una adecuada ingesta dietética. En la circulación, la cantidad de calcio y magnesio es menos del 1% del contenido total del organismo. Sin embargo, alteraciones en las concentraciones séricas de estos minerales pueden comportar alteraciones de funciones fisiológicas importantes, con una sintomatología muy variada. En esta revisión se presenta un breve resumen de la homeostasis que permitirá entender mejor las posibles causas que pueden alterar ésta, las consecuencias que pueden derivar y su tratamiento.
Puntos clave

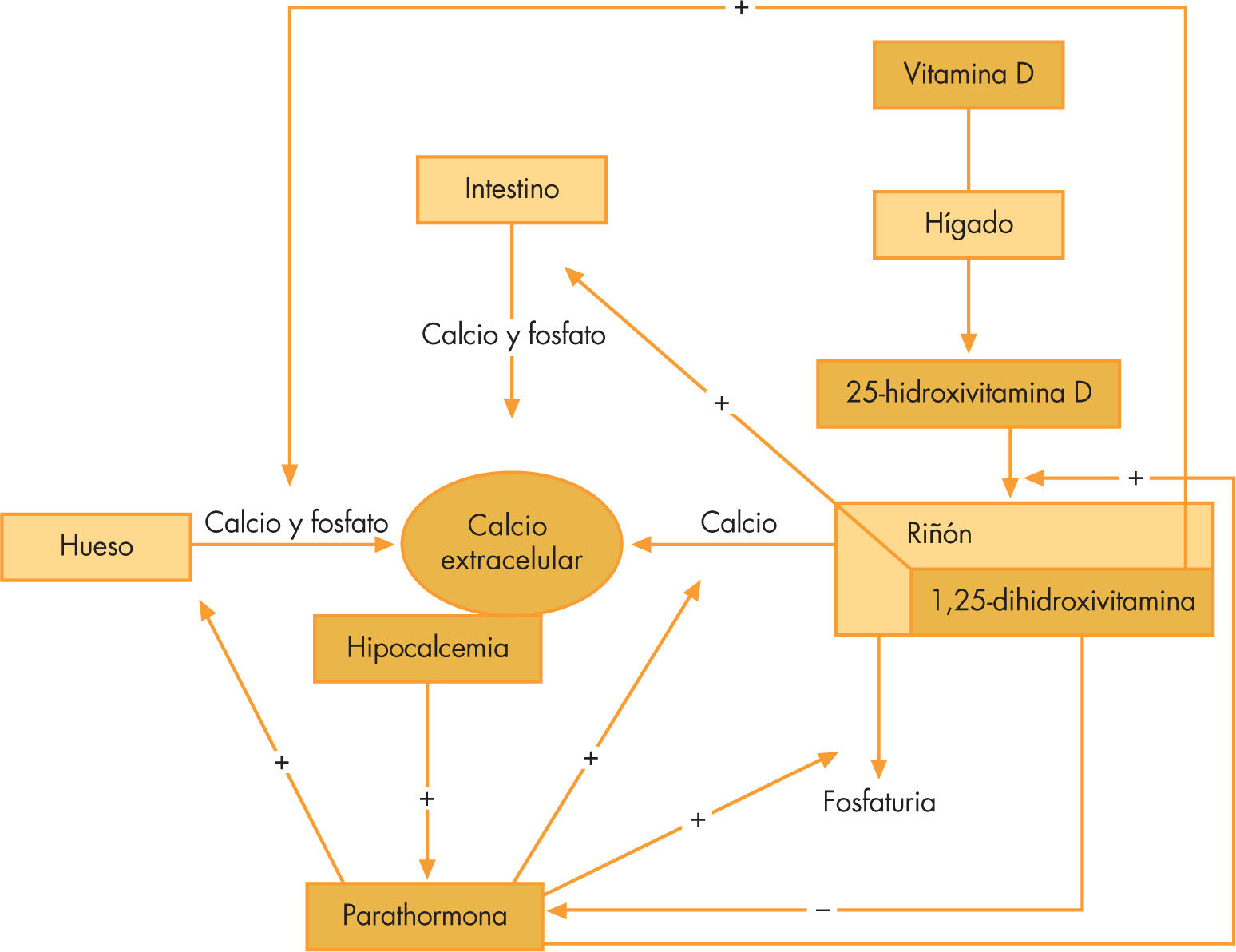
El 98% del calcio total se encuentra en el hueso. Del resto dependen importantes funciones intracelulares, como la transmisión de señales y numerosas reacciones enzimáticas y extracelulares, como la coagulación, la secreción endocrina, la conducción nerviosa y la contractibilidad muscular. El calcio sérico se encuentra en 3 formas: unida a proteínas, principalmente a la albúmina (30-50%); formando complejos con aniones como el fosfato, citrato o bicarbonato (5-15%), e ionizado (40-60%). La forma ionizada (Ca2+) es la metabólicamente activa y es la fracción soluble. Esta pequeña fracción depende de la absorción de calcio en el intestino, del continuo recambio mineral del hueso y del balance renal de este ion. Para ello, paratohormona (PTH), vitamina D y, en menor grado, calcitonina regulan este equilibrio actuando sobre sus dianas: intestino, hueso y riñón (fig. 1).

La calcemia total normal oscila entre 8,7 y 10,8mg/dl y la iónica entre 4,4 y 5,4mg/ dl (1,1 y 1,35mmol/l). Si no se dispone del calcio iónico hay que tener en cuenta los factores que influencian al calcio sérico1. Las variaciones del pH sanguíneo modifican la unión calcio-albúmina: cada 0,1 unidad que baja el pH, aumenta 0,03mmol/l el calcio iónico. Por otro lado, el calcio iónico depende del valor de albúmina, por lo que si no se dispone de la calcemia iónica debe calcularse ésta teniendo en cuenta la calcemia total y la albuminemia con la siguiente fórmula: Ca (mg/dl) = Ca medido + 0,8 (4 - albúmina [mg/dl]).
El receptor sensor de calcio presente en las células paratiroideas es un “calciostato” que ajusta el 50% de la secreción máxima de PTH a la calcemia iónica normal (Ca2+ 1,25mmol/l). Así, cambios en la calcemia regulan de forma rápida la secreción de PTH. Reducciones y elevaciones del valor de calcemia están asociados con un aumento y una disminución de la PTH, respectivamente2–6. Estos receptores de calcio también se expresan en los riñones, así que el calcio iónico ejerce directamente una regulación en su absorción en el túbulo distal (la hipercalcemia inhibe la reabsorción)7. Además de la hipocalcemia, la hiperfosfatemia aumenta la secreción de PTH. Sin embargo, la forma activa de la vitamina D, el calcitriol, y la hipo e hipermagnesemia inhiben la secreción de PTH8,9. En el hueso, la PTH promueve el recambio óseo, facilitando la salida de calcio y fosfato desde el hueso. En el riñón, estimula la reabsorción de calcio y la eliminación de fosfato. Además, la PTH estimula la secreción de calcitriol (forma activa de la vitamina D). Por último, en el intestino, estimula la absorción calcio y fosfato a través de la acción de calcitriol. En resumen, de forma neta, la PTH aumenta la calcemia sin afectar de forma considerable los valores de fosfatemia.
La vitamina D3 (colecalciferol) es el producto de la fotolisis del 7-dehidrocolesterol bajo la radiación ultravioleta en la piel10. Ésta se hidroxila a 25-hidroxivitamina D en el hígado, y a 1,25-dihidroxivitamina D (o calcitriol) en el túbulo renal proximal. La PTH, la hipocalcemia y la hipofosfatemia son los mayores inductores de la 1-α- hidroxilasa. Esta enzima se expresa también en células mononucleares y podría ser la responsable del aumento de vitamina D e hipercalcemia presente en algunos desórdenes granulomatosos, como la sarcoidosis11. En el hueso y el intestino, la vitamina D promueve el recambio óseo y la absorción intestinal de calcio y fosfato, respectivamente. En el riñón facilita la acción de la PTH. El resultado neto del calcitriol es que aumenta la calcemia y la fosfatemia.
La calcitonina se produce por las células C parafoliculares del tiroides. La hipercalcemia activa su secreción y tiene efectos contrarios a la PTH en hueso y riñón, sin tener ninguna acción en el intestino.
HipocalcemiaEn el sistema nervioso, la hipocalcemia puede producir parestesias periorales y en la yema de los dedos, hiperreflexia, calambres musculares, espasmo carpopedal, convulsiones, alteración del sensorio y tetania (la latente puede ponerse de manifiesto mediante los signos de Chovsteck y Trousseau). Por otro lado, puede provocar arritmias cardíacas, hipotensión e insuficiencia cardíaca. La hipocalcemia crónica puede producir papiledema, neuritis óptica, catarata subcapsular, sequedad de piel, aspereza del cabello, fragilidad de uñas, hipoplasia dentaria y calcificación de los ganglios basales. En el neonato, puede haber irritabilidad, vómitos, rechazo de la ingesta, fasciculaciones, convulsiones, apneas o hipotonía.
Lectura rápida

La forma ionizada del calcio sérico es la foma metabólicamente activa. Si no se dispone del calcio iónico, hay que tener en cuenta que alteraciones del pH y del valor de albúmina pueden simular situaciones de seudohipocalcemia. Su correcta homeostasis depende del balance entre una ingesta adecuada, el valor de hormona paratiroidea, vitamina D, y de su balance entre la absorción intestinal, el recambio óseo y la absorción renal. De forma resumida, la PTH aumenta la calcemia y disminuye la fosfatemia, mientras que la vitamina D aumenta calcemia y la fosfatemia.
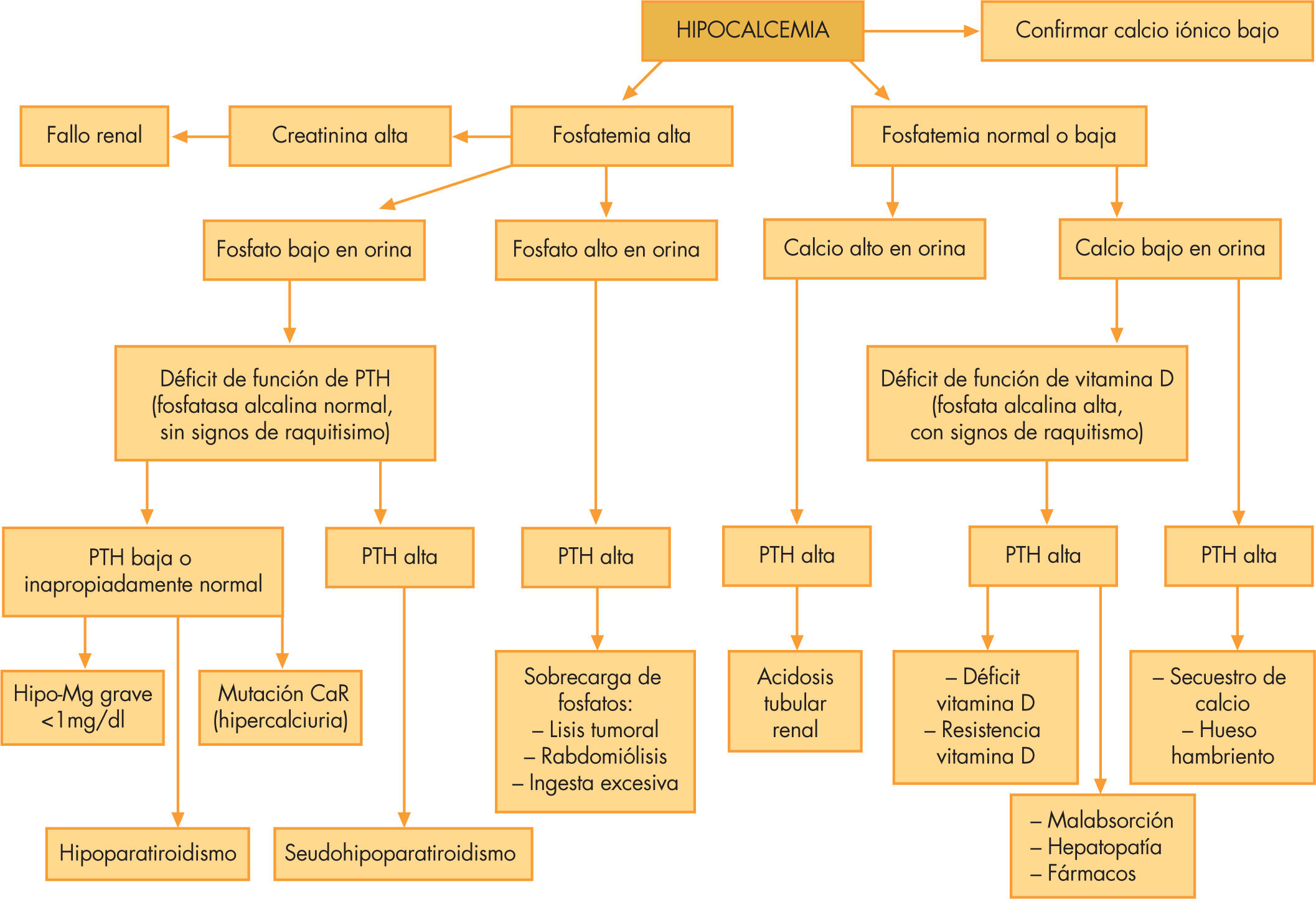
HipocalcemiaAnte una hipocalcemia, es importante determinar la cifra de fosfato puesto que nos orientará hacia la causa subyacente. Una hiperfosfatemia debe hacer pensar en un hipoparatiroidismo o seudohipoparatiroidismo, mientras que un fosfato no elevado en un contexto de hipocalcemia, por su frecuencia, debería orientarnos hacia un déficit de vitamina D.

La etiología de la hipocalcemia es muy amplia. Ante una hipocalcemia, la cifra de fosfato nos orientará hacia la causa aún sin disponer de ningún resultado hormonal (fig. 2). Hipocalcemias con hiperfosfatemias deben hacer pensar en un hipoparatiroidismo o seudohipoparatiroidismo (por la acción fosfatúrica de esta hormona). Sin embargo, si el fosfato no está elevado habrá que pensar en el resto de causas, entre las que destaca el déficit de vitamina D.

Lectura rápida
La hipocalcemia sintomática debe tratarse de forma urgente con gluconato cálcico por vía intravenosa, y en caso de acompañarse de una hipomagnesemia ésta debe ser corregida también. En tratamientos crónicos, debe mantenerse un valor de calcemia que mantenga al paciente libre de síntomas, pero procurando evitar algunas complicaciones que pueden darse con esta terapia como son la nefrocalcinosis y los depósitos de fosfato cálcico en tejidos. Según la etiología de la hipocalcemia, hay que recordar añadir suplementos de ergocalciferol o calcitriol.
HipercalcemiaEn cuanto a las causas de hipercalcemia, hay que recordar el hiperparatiroidismo, ya sea primario (por un adenoma o hiperplasia) o secundario a otra causa de hipocalcemia que haya estimulado la producción de hormona paratiroidea (PTH). Además del aumento de la PTH, el exceso de vitamina D (por incremento de la ingesta o asociada a procesos granulomatosos o enfermedades neoplásicas) puede producir un aumento del calcio plasmático. Los estados de inmovilización, el hipertiroidismo y las metástasis óseas pueden conducir también a situaciones de hipercalcemia, dado que favorecen el recambio óseo.
En primer lugar, hay múltiples causas de hipoparatiroidismo que se acompañarán de hipocalcemia, hiperfosfatemia y PTH baja. Dentro de las formas congénitas hay formas familiares de hipoparatiroidismo (ya sean de herencia autosómica dominante o recesiva o ligadas al cromosoma X) en las que se produce una PTH inactiva o una PTH activa12, pero de expresión disminuida. El síndrome de DiGeorge es otra enfermedad en la que hay una agenesia o disfunción congénita de la paratiroides asociada a defectos cardíacos, dimorfismo facial y susceptibilidad para las infecciones. Por otro lado, ciertos pacientes con hipocalcemia autosómica dominante presentan mutaciones de los receptores paratiroideos sensores del calcio, produciendo una inhibición de la secreción de la PTH con valores de calcemia más bajos de lo normal13.
El hipoparatiroidismo adquirido puede deberse a enfermedades infiltrativas o de depósito, como la enfermedad de Wilson o procesos granulomatosos, a enfermedades autoinmunitarias que provocan la destrucción de la glándula (enfermedad poliglandular autoinmune tipo I), o radioterapia o cirugía de ésta. También se observa una secreción anormal de PTH en enfermos críticos y en la hipomagnesemia.
En segundo lugar, la hipocalcemia puede deberse a un seudohipoparatiroidismo (con hiperfosfatemia y PTH elevada), en la que hay una resistencia a la PTH causada por la mutación de factores que se requieren en la transducción de señales intracelulares mediada por el receptor de la PTH.
En tercer lugar, el origen de la hipocalcemia puede ser una inadecuada acción de la vitamina D, encontrando hipocalcemia, fosfatemia variable con un valor de PTH adecuado. Se puede deber a un déficit de vitamina D14, con valores disminuidos (por ingesta escasa, patología hepática que no permite que se produzca la 25-hidroxilación o por fármacos que metabolizan la vitamina D a formas inactivas) o por una resistencia a la vitamina D, con valores aumentados (ya sea por déficit de la 1-α-hidroxilasa o por receptores alterados del calcitriol).
En cuarto lugar, alteraciones de los órganos involucrados en la homeostasis de la calcemia pueden causar hipocalcemia. Así ocurre en la insuficiencia renal, la acidosis tubular renal, las patologías con malabsorción intestinal o el llamado síndrome del hueso hambriento (hipocalcemia que se produce tras una paratiroidectomía por un hiperparatiroidismo que ha estado generando una salida de calcio desde el hueso al plasma y que ha dejado al hueso “hambriento de calcio”).
Otras causas de hipocalcemia son la hiperfosfatemia por síndrome de lisis tumoral, rabdomiólisis o alta ingesta de fosfatos; el secuestro de calcio por ácidos grasos libres en la pancreatitis o por la presencia de citrato en las exanguinotransfusiones, y fármacos como la furosemida por su capacidad calciúrica, y la calcitonina y los bisfosfonatos por la inhibición del recambio óseo.
El tratamiento de la hipocalcemia del paciente sintomático debe realizarse con una perfusión intravenosa de gluconato cálcico al 10% (9,3mg Ca/ml) a 0,2ml/kg en más de 10min para evitar alteraciones del ritmo cardíaco con monitorización del electrocardiograma. Posteriormente, se debe mantener la normocalcemia con aportes de calcio elemental de 20–80mg/kg/día de forma continua (preferiblemente ya que los bolos se asocian a una mayor pérdida de calcio por orina) o en bolos cada 4–6h. Si el paciente está asintomático y la hipocalcemia no es grave, es mejor la vía oral (50–100mg/kg/día) cada 4–6h en forma de carbonato cálcico (40% calcio), citrato cálcico (21% calcio), lactato cálcico (13% calcio), gluconato cálcico (9,4% calcio) o glucobionato cálcico (6,6% calcio). Se debe intentar conseguir un valor de calcio que no produzca síntomas al niño evitando la hipercalcemia, la excesiva hipercalciuria (mantener índice urinario calcio/creatinina < 0,2 para evitar la nefrocalcinosis) y los depósitos en articulaciones o tejido blando de fosfato cálcico (mantener un producto de calcio × fosfato < 70).
La hipomagnesemia, que por si sola puede inhibir la secreción y acción de la PTH, debe ser corregida mediante MgSO4 (50% solución) en el momento agudo con 25–50mgMg2+/kg/dosis cada 4–6h (por vía intravenosa o intramuscular). Posteriormente debe mantenerse una dosis de 30–60mgMg2+/kg/día por vía intravenosa u oral.
Por otro lado, debe administrarse calcitriol a dosis de 0,01-0,05μg /kg/día en los casos de hipoparatiroidismo, seudohipoparatiroidismo y enfermedad renal o hepática. En el caso de déficit de vitamina D, el tratamiento con calcitriol no es necesario ya que no hay un defecto en la producción de la forma activa de la vitamina D, por lo que con ergocalciferol por vía oral en dosis de 1.200 a 1.600 unidades al día, o una dosis única intramuscular de 50.000 o 100.000 unidades, sería suficiente.
HipercalcemiaLa hipercalcemia en el sistema nervioso central puede provocar encefalopatía y convulsiones. Al igual que la hipocalcemia provoca arritmias cardíacas y, en este caso, hipertensión. Dado que provoca una no respuesta renal de la hormona antidiurética, la hipercalcemia puede originar poliuria. También puede ocasionar estreñimiento, anorexia, vómitos y calcificaciones en piel y órganos. En el neonato, la forma de presentación puede ser hipotonía, apnea y distrés respiratorio.
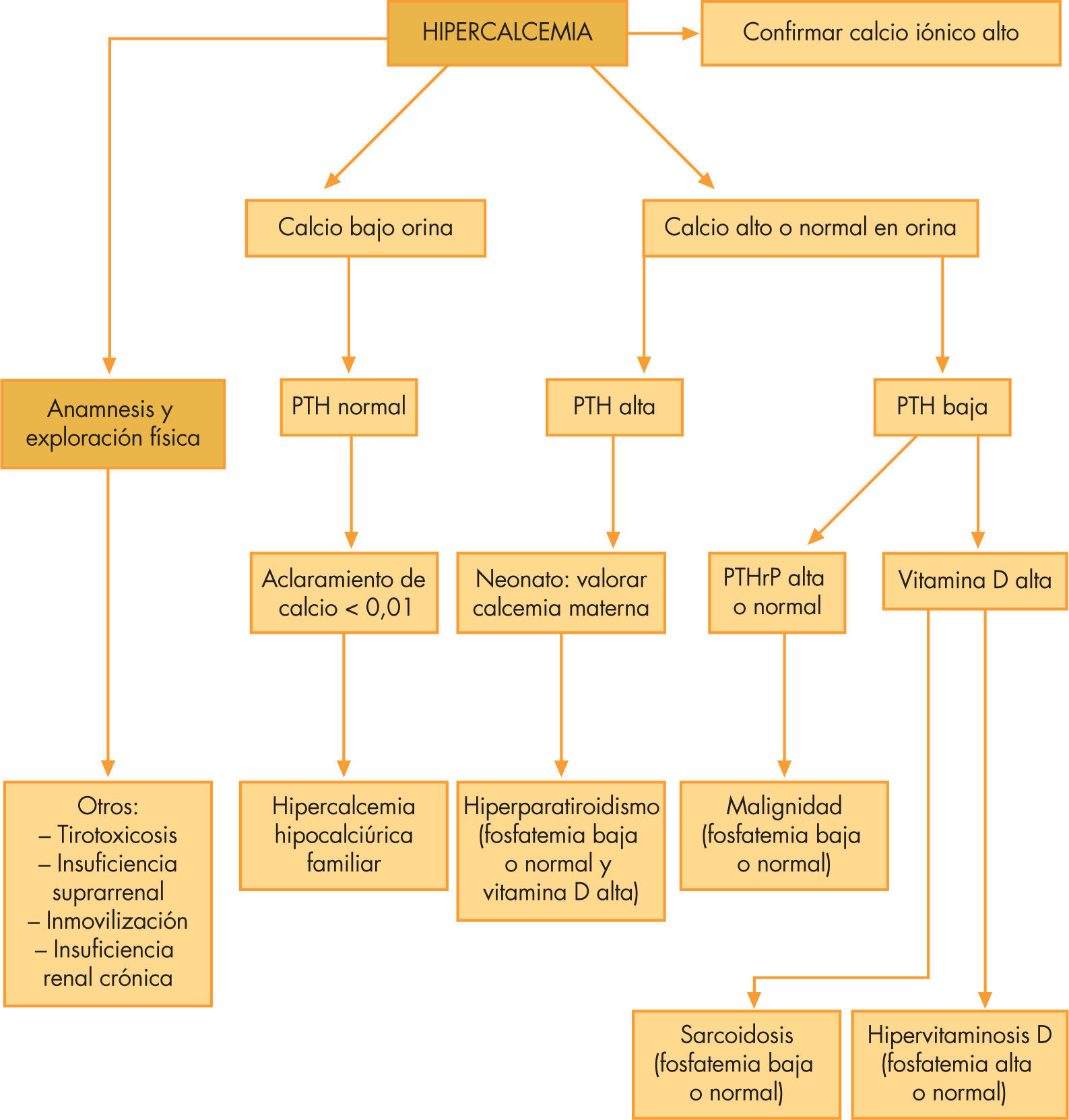
En cuanto a la etiología, en primer lugar se hablará del hiperparatiroidismo (hipercalcemia, fosfato normal o bajo, PTH alta y calcitriol elevado). El hiperparatiroidismo primario puede presentarse por hiperplasia de la glándula paratiroidea o por un adenoma, y puede ser una presentación esporádica o asociada a otras neoplasias endocrinas (MEN tipo 1 y 2A). Además del primario, existe el hiperparatiroidismo secundario (ocasionado por una hipocalcemia originada por otra causa, como la insuficiencia renal o el déficit de vitamina D) y el terciario (cuando la hiperplasia paratiroidea del hiperparatiroidismo secundario se convierte en autónoma y deja de compensar una hipocalcemia de otra causa). Por otro lado, en el período neonatal el niño puede tener un hiperparatiroidismo transitorio secundario a un hipoparatiroidismo materno.
Lectura rápida
El tratamiento de la hipercalcemia consiste en hiperhidratar al paciente y asociar un diurético calciúrico como la furosemida. En hipercalcemias rebeldes al tratamiento puede utilizarse calcitonina, y en los estados en los que hay un exceso de vitamina D el uso de glucocorticoides puede ser efectivo, dado que inhibe la 1-α-hidroxilasa y la absorción intestinal.
Mención aparte se debe hacer de la hipercalcemia hipocalciúrica familiar (hipercalcemia, con un valor de PTH inapropiadamente normal o elevado y una fosfatemia baja o normal), de herencia autosómica dominante, causada por una mutación en el receptor de calcio paratiroideo que lo hace menos sensible, de manera que se requieren valores mayores de calcemia de lo habitual para inhibir la PTH15. Habitualmente, se caracteriza por una moderada hipercalcemia asintomática sin los hallazgos esqueléticos típicos del hiperparatiroidismo primario y con menor riesgo de litiasis renal, debido a hipocalciuria. Sin embargo, el espectro de presentación es muy amplio, y se puede presentar como una forma grave de hipercalcemia neonatal y todas las manifestaciones del hiperparatiroidismo primario.
Una situación que simula un hiperparatiroidismo (hipercalcemia, hipofosfatemia, pero con PTH baja y vitamina D normal), es la secreción excesiva de PTHrP (paratohormone related peptide) que ejerce la mayoría de las funciones biológicas de la PTH a través del mismo receptor. La PTHrP puede ser producida por varios tumores de estirpe epitelial.
En segundo lugar, el exceso de vitamina D por excesiva ingesta puede producir hipercalcemia (hiperfosfatemia y PTH baja. En situaciones con una proliferación y activación de células monocíticas (como la sarcoidosis o algunas neoplasias) la producción de calcitriol se ve aumentada debido a una disregulación de la expresión de la 1-α-hidroxilasa en estas células.
En los huesos, la inmovilización promueve la hipercalcemia, así como las metástasis óseas y el hipertiroidismo16 dado que el exceso de hormona tiroidea estimula la acción de los osteoclastos.
La ingesta excesiva de calcio o el déficit de ingesta de fosfatos pueden conducir a una hipercalcemia, así como el tratamiento con tiazidas (por reabsorción de calcio a nivel renal), con vitamina A17 (por activación de los osteoclastos) o con litio (porque disminuye la sensibilidad de los receptores de calcio de la paratiroides).
Hay otras causas de hipercalcemia de etiología aún no bien definidas como son: el síndrome de Williams, la hipofosfatasia, la necrosis grasa subcutánea, la insuficiencia suprarrenal o el feocromocitoma. En la figura 3 se propone un algoritmo para ayudar a orientar la causa de una hipercalcemia.

La primera línea de terapia, además de la etiológica, debería ser hiperhidratar y aumentar la excreción de calcio urinario. Los pacientes sintomáticos deben hidratarse con suero fisiológico a 3.000ml/m2 en las primeras 24–48h. Después de la hidratación, administrar furosemida a 1mg/kg cada 6h. En los casos graves (p. ej., en pacientes comatosos), se debe considerar la hemodiálisis.
Cuando la hipercalcemia no responde a estas medidas o cuando se debe a un aumento del recambio óseo, puede usarse la calcitonina (4 U/kg intravenosa cada 12h), aunque su empleo de forma continuada disminuye su efecto por taquifilaxia. En casos de hipercalcemia por movilización de calcio desde el hueso, ya sea por malignidad, hiperparatiroidismo grave o inmovilización, se han usado bisfosfonatos, pero la experiencia del efecto de estos fármacos en el crecimiento en niños es limitado. El etidronato a 7,5mg/kg/día y el pamidronato a 0,5-1mg/kg/dosis (dosis única intravenosa) pueden ser de utilidad.
En los casos causados por aumento de vitamina D, los glucocorticoides (prednisona 1mg/kg/día) puede ser efectiva ya que inhibe la 1-α-hidroxilasa y la absorción intestinal de calcio.
Lectura rápida
En las situaciones de hipofosfatemia, la cifra de fosfato en orina nos ayudará a esclarecer la etiología del cuadro. En el hiperparatiroidismo, el raquitismo hipofosfatémico o el síndrome de Fanconi, la cifra de fosfato en orina estará aumentada. Sin embargo, cuando la hipofosfatemia se deba a un déficit de la ingesta o a problemas malabsortivos intestinales el fosfato en orina estará disminuido. Por otro lado, hay que recordar que durante el tratamiento de la cetoacidosis diabética, no es extraño encontrar una hipofosfatemia. El tratamiento de la hipofosfatemia debe realizarse con suplementos de fósforo elemental, asociando, según la causa que la origine y siempre que sea necesario, aportes de vitamina D.
El fósforo se encuentra en forma de fosfatos orgánicos (ATP, creatín fosfato, glucofosfatos, fosfolípidos) de gran importancia metabólica y de fosfatos inorgánicos (Pi) en el esqueleto, siendo básicos para la cristalización de la hidroxiapatita y un eficaz mecanismo para la excreción renal de hidrogeniones. En una situación estable, los valores de fósforo están condicionados principalmente por la capacidad renal para excretar el fósforo de la dieta. Mínimas elevaciones de la fosforemia tras la ingesta condicionan una inhibición del cotransporte sodio-fósforo en el túbulo proximal y escape de éste. La vitamina D y la PTH participan también, como se ha visto anteriormente, en la regulación metabólica del fósforo.
HipofosfatemiaLa hipofosfatemia aguda es usualmente asintomática, pero su déficit crónico puede provocar alteración de la liberación del oxígeno por la hemoglobina porque disminuye el 2,3 difosfoglicerato en los eritrocitos, pudiendo ocasionar hipoxia cerebral. También puede provocar debilidad muscular, insuficiencia respiratoria, miocardiopatía, desmineralización y deformidades óseas.
La hipofosfatemia puede producirse por depleción de fósforo por pérdida renal, por disminución de la absorción intestinal de fósforo y por redistribución del fósforo extracelular. En las situaciones con un aumento de la PTH, el efecto fosfatúrico de esta hormona condiciona una hipofosforemia. En el raquitismo hipofosfatémico y el síndrome de Fanconi también se observa una hiperfosfaturia que conduce a una hipofosfatemia. Por otro lado, no hay que olvidar el efecto fosfatúrico de determinados fármacos diuréticos.
En segundo lugar, la hipofosfatemia se puede deber a un aporte escaso de fosfatos (p. ej., en neonatos pretérmino alimentados con leche artificial no suplementada18, o por escaso aporte en la nutrición parenteral19). El déficit de vitamina D20 o las enfermedades malabsortivas pueden ser la causa de una disminución de la absorción intestinal de fósforo. En estos casos, por supuesto, el fosfato en orina estará disminuido.
Por último, la redistribución del fósforo extracelular puede generar una hipofosforemia21. Con la glucólisis intracelular se forman carbohidratos fosforilados a través de un proceso estimulado por insulina. Al ser el fósforo inorgánico extracelular la fuente del intracelular, si la glucólisis está estimulada puede aparecer hipofosforemia. Esto ocurre en el tratamiento de la cetoacidosis diabética, en la hiperglucemia no cetósica o durante la administración de carbohidratos en el proceso de realimentación de pacientes malnutridos. En las situaciones con alcalosis respiratoria se estimula la fosfofructocinasa intracelular, que a su vez estimula la glucólisis, lo que puede provocar también hipofosforemia. Por último, en el síndrome del hueso hambriento, además de hipocalcemia, puede aparecer hipofosfatemia.
La hipofosfatemia de origen nutricional se debe tratar con suplementos de fósforo elemental (0,5 a 1,5mmol/kg/día). Puede ser útil añadir también suplementos de calcio para minimizar la hipocalcemia producida durante el tratamiento de la hipofosforemia. Hay que recordar que en los casos producidos por déficit de vitamina D deberán pautarse aportes de ésta. En los casos en los que el origen de la hipofosfatemia no sea nutricional, el tratamiento etiológico será primordial para conseguir mantener un valor de fósforo normal.
Lectura rápida
En el caso de estar ante una hiperfosfatemia, poder determinar la pérdida renal de fosfato por orina también orientará nuestro diagnóstico. En caso de insuficiencia renal, hipoparatiroidismo o seudohipoparatiroidismo, la pérdida de fosfato por orina estará disminuida. Sin embargo, si la hiperfosfatemia se debe a una ingesta excesiva con la dieta o a situaciones de rabdomiólisis o lisis tumoral, el fosfato en orina estará aumentado. El manejo de la hiperfosfatemia aguda se basa en la hiperhidratación con suero fisiológico y la terapia farmacológica con acetazolamida.

La hiperfosfatemia puede ser asintomática o manifestarse por los síntomas de hipocalcemia que pueden acompañarle.
La hiperfosfatemia22 puede producirse por una disminución de la excreción renal de fosfato, por una sobrecarga de fósforo o por un movimiento transcelular de éste. Si nos encontramos ante una hiperfosfatemia con pérdida de fosfato en orina disminuida, debemos pensar que la causa se encuentra en un problema renal o en un hipoparatiroidismo. En el resto de causas, la excreción de fosfato por la orina debería estar aumentada.
La insuficiencia renal aguda o crónica debida a la disminución de la filtración y excreción del fosfato es la causa más frecuente. Inicialmente, la acción de la PTH aumentada compensa la situación dado su acción fosfatúrica, pero la retención de fósforo empieza cuando el filtrado glomerular cae por debajo de 30ml/min. Otras causas de hiperfosfatemia por un aumento de la reabsorción tubular son el hipoparatiroidismo o seudohipoparatiroidismo, la acromegalia23 (la hormona de crecimiento podría aumentar la reabsorción), el tratamiento con bisfosfonatos24 (estimulación directa de la reabsorción), la hipervitaminosis D y la calcinosis familiar tumoral25 (enfermedad autosómica recesiva que se caracteriza por hiperfosfatemia debida a un aumento de la reabsorción renal, asociada generalmente a una concentración de calcitriol sérica aumentada).
En segundo lugar, la sobrecarga de fósforo puede ser por el aporte excesivo de éste en la nutrición, ya sea por ingerir leche o cereales con alto contenido de fósforo o por aportes elevados mediante nutrición parenteral. Además, la administración de laxantes que contienen sales de fósforo puede suponer una sobrecarga de éste. La hipervitaminosis D o A también puede ocasionar hiperfosfatemia dado que aumenta el recambio óseo. Por último, hay algunas situaciones en las que se produce una hiperfosfatemia debido a la movilización de fosfato desde el espacio intracelular al extracelular. Este hecho se produce en la acidosis láctica o en la cetoacidosis diabética, en el síndrome de lisis tumoral (por el efecto de determinados fármacos citotóxicos en algunos tumores con alto recambio celular) y en la rabdomiólisis.
Si la función renal es normal, la hiperfosfatemia aguda se trata forzando la diuresis con suero fisiológico (véase tratamiento de hipercalcemia) y, en casos refractarios, puede probarse la terapia con acetazolamida. Si la función renal está muy alterada y el paciente se encuentra sintomático, tiene que considerarse la conveniencia de realizar hemodiálisis. En los casos con hiperfosfatemia crónica, el tratamiento consiste en restringir el fósforo de la dieta. El uso de calcio o sales de aluminio como quelante del fosfato intestinal en presencia de hipofosfatemia puede predisponer a la formación de calcificaciones metastásicas26 o a una toxicidad potencial por aluminio27, respectivamente, por lo que el primero debe utilizarse con precaución y las sales de aluminio no están recomendadas28,29.
