














La fibrosis quística (FQ) es una enfermedad hereditaria de carácter autosómico recesivo debida a un defecto en el transporte epitelial de electrolitos que conduce, como hallazgos más significativos, a la producción de una enfermedad pulmonar supurativa crónica y al desarrollo de una insuficiencia pancreática crónica con malabsorción digestiva.
En las últimas 2 décadas, se ha profundizado enormemente en el conocimiento de su etiología y patogenia, y en el desarrollo de nuevas medidas terapéuticas, lo que ha permitido mejorar la supervivencia y conseguir que la enfermedad deje de pertenecer exclusivamente al campo de la pediatría. En la actualidad, se desarrollan nuevos métodos terapéuticos, como el tratamiento génico o el tratamiento molecular, al igual que se refinan los tratamientos actuales con la síntesis de nuevos antibióticos, mucolíticos, suplementos pancreáticos, preparados vitamínicos y mejores productos nutricionales.
En el presente trabajo, se hará una revisión sobre los conceptos actuales de la clínica y el tratamiento de los pacientes con FQ desde el punto de vista digestivo y nutricional, y se finalizará con una visión de los nuevos tratamientos en desarrollo en el momento actual.
Puntos clave

La frecuencia de presentación de la enfermedad depende de la población estudiada. En población blanca, es de 1 por cada 1.500 recién nacidos vivos, lo que supone una frecuencia de 1 portador por cada 20–25 sanos1.
En otras razas, su incidencia es mucho menor: 1 de cada 17.000 recién nacidos vivos en población afroamericana y 1 de cada 90.000 en países de Extremo Oriente2,3.
EtiopatogeniaLa FQ está causada por una mutación en el brazo largo del cromosoma 7 (7q31) en el gen que codifica la proteína denominada CFTR (del inglés cystic fibrosis transmembrane conductance regulator)4,5. El CFTR pertenece a un grupo de proteínas fijadoras de adenosintrifosfato y reguladas por adenosinmonofosfato cíclico que actúan como transportadores epiteliales de cloro negativo y bicarbonato. De manera adicional, influye también en la composición de la interfase líquida de la superficie de las vías respiratorias, a través de la interacción con otras proteínas, fundamentalmente canales de sodio. El resultado de su ausencia o disfunción es una disregulación del contenido hidroelectrolítico en los epitelios en los que se expresa esta proteína (pulmón, páncreas, intestino, tracto hepatobiliar, glándulas sudoríparas y vasos deferentes), lo que da lugar a la producción de un moco hiperviscoso que condiciona la mayoría de las manifestaciones de la enfermedad.
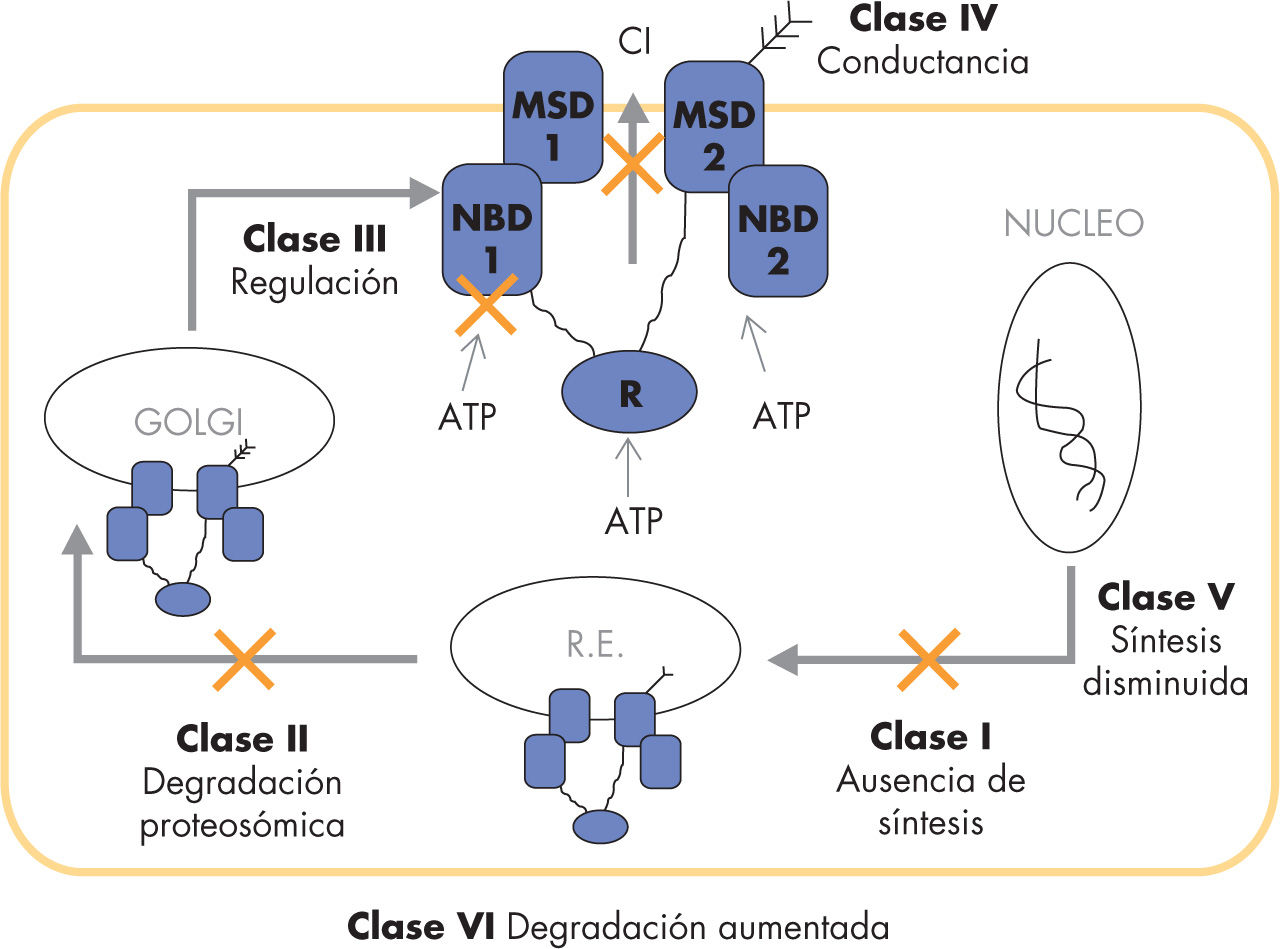
En la actualidad, se han descrito más de 1.200 mutaciones capaces de producir la enfermedad, las cuales se clasifican en 6 tipos (fig. 1)6:
- 1.
Clase I o déficit de síntesis. Hay una interrupción en la traducción del ácido desoxirribonucleico (ADN) a ácido ribonucleico mensajero (ARNm), generalmente debido a mutaciones sin sentido o a codones de interrupción temprana.
- 2.
Clase II o de degradación proteosómica. Alteraciones del plegamiento proteico impiden su procesamiento en el retículo endoplásmico y Golgi, con lo que el CFTR no alcanza la membrana citoplasmática.
- 3.
Clases III (alteraciones de la regulación) y IV (alteraciones de conductancia). El CFTR completo alcanza la membrana celular, pero su función está disminuida.
- 4.
Clase V. Hay una menor traducción de ADN en el ámbito nuclear, por lo que se produce CFTR en cantidades menores.
- 5.
Clase VI. Tiene lugar una disminución de la vida media del CFTR en la membrana, con una disminución global del número de transportadores.
, los defectos por alteraciones de maduración y degradación proteosómica (clase II), las alteraciones de la regulación (clase III) y de la conductancia del canal (clase IV) y la reducción del número de transportadores debido a una menor síntesis (clase V) o a una mayor degradación (clase VI). ATP: adenosintrifosfato; Cl: cloro; MSD: dominio de fijación de la membrana (membrane spanning domain); NBD: dominio fijador de nucleótidos (nucleotide binding domain); R: dominio regulador.")
Clasificación de las mutaciones del genCFTR. Los defectos del gen CFTR incluyen la ausencia de síntesis (clase I), los defectos por alteraciones de maduración y degradación proteosómica (clase II), las alteraciones de la regulación (clase III) y de la conductancia del canal (clase IV) y la reducción del número de transportadores debido a una menor síntesis (clase V) o a una mayor degradación (clase VI). ATP: adenosintrifosfato; Cl: cloro; MSD: dominio de fijación de la membrana (membrane spanning domain); NBD: dominio fijador de nucleótidos (nucleotide binding domain); R: dominio regulador.
La frecuencia de cada mutación varía según la población estudiada, siendo las más habituales las de clase II. En nuestro medio, las 2 más frecuentes son de la ΔF508 (clase II) y la G542X (clase I), presentes en alrededor del 70% de los pacientes6. Se ha intentado determinar en qué medida influye el genotipo en la expresión fenotípica de la enfermedad, aunque los resultados obtenidos han sido poco determinantes en este aspecto. En general, los homozigotos para las mutaciones de clases I, II y III tendrían peor pronóstico, con medianas de supervivencia menores que los homozigotos o heterozigotos para las de clases IV a VI7. Sin embargo, esto suele cumplirse más en relación con la afectación pancreática que con la pulmonar, y es de poca utilidad para valorar el pronóstico de cada paciente de forma individual.
En cualquier caso, la disfunción del CFTR produce, a nivel pancreático, unas secreciones viscosas, deficitarias en agua y bicarbonato, que forman fácilmente tapones en los ductos intralobulares y conducen a la digestión retrógrada de la glándula, con desaparición de los acini, que se reemplazan por tejido fibroso que rodea zonas quísticas (lo que da nombre a la enfermedad), y con el desarrollo consiguiente de una insuficiencia pancreática exocrina que, a la larga, también puede ser endocrina por destrucción de los islotes pancreáticos.
En el ámbito intestinal, la retención mucosa en las glándulas de Brunner y las células caliciformes facilita la producción de secreciones intestinales espesas y viscosas que justifican algunas manifestaciones de la FQ como el íleo meconial o el síndrome de obstrucción intestinal distal (SOID).
Por último, la expresión en vías biliares de un CFTR defectuoso favorece la producción de una bilis menos fluida y alcalina, debido a la menor secreción de agua y bicarbonato, lo que dificulta el flujo biliar ductular y aumenta la susceptibilidad del epitelio biliar a lesionarse, debido a la acción detergente de ácidos biliares endógenos y a la presencia de agentes infecciosos.
Manifestaciones clínicas de comienzoAunque hay una forma clásica, en la práctica la enfermedad puede iniciarse en momentos y de formas diversas, abarcando desde el período intraútero o neonatal inmediato hasta etapas más tardías de la infancia o, incluso, de la vida adulta. Así, la FQ puede iniciar con cualquiera de los síntomas característicos de ésta8.
Ileo meconialEntre un 10 y un 20% de los pacientes presentan en el período neonatal inmediato un cuadro de obstrucción intestinal debido a la acumulación de meconio espeso en la zona del íleon distal. Esta obstrucción, de comienzo intrauteral, es casi exclusiva de pacientes con insuficiencia pancreática, y se desconoce por qué sólo se presenta en una pequeña parte de éstos. En algunos casos puede producirse una perforación intestinal durante la gestación, dando lugar a una peritonitis meconial, y posteriormente se observa la presencia de calcificaciones en la radiografía simple de abdomen. De manera característica, la ausencia de tránsito distal a la obstrucción produce un microcolon por falta de uso, que es reversible una vez reinstaurada la continuidad del tránsito intestinal.
En algunos casos, el cuadro puede no ser tan dramático, dando lugar a retrasos o a dificultad en la evacuación de meconio.
El tratamiento de estos recién nacidos dependerá de la intensidad del cuadro y de la existencia o no de complicaciones quirúrgicas, como perforación o asociación de otras malformaciones, como atresias de intestino delgado. En cualquier caso, el diagnóstico de íleo meconial obliga en todos los casos a investigar la presencia de una FQ3.
Forma clásica de la enfermedadAlrededor del 85% de los pacientes presenta insuficiencia pancreática, que puede ser evidente desde el nacimiento o desarrollarse durante el primer año de vida.
Se produce una diarrea crónica secundaria a la malabsorción de grasa y proteínas, con deposiciones típicas de características esteatorreicas: abundantes, no muy numerosas, de aspecto pálido, brillante y adherente y de olor rancio. La consecuencia es una malnutrición calóricoproteica progresiva (a la que contribuyen otros factores, como el aumento del gasto energético por la presencia de la afectación respiratoria y la disminución de la ingesta), junto con una pérdida progresiva de vitaminas liposolubles, cuya absorción se ve disminuida en relación con la malabsorción de grasa.
Este cuadro de diarrea prolongada durante el primer año de vida, con fracaso del desarrollo y la malnutrición, acompañado de la afectación respiratoria con infecciones frecuentes y de evolución tórpida, constituye la forma clásica de presentación de la enfermedad8.
Presentación con pérdida hidrosalinaLa disfunción del CFTR en el conducto de la glándula sudorípara produce un defecto en la resorción de cloro negativo en el canal, con la consiguiente producción de un sudor con un mayor contenido en cloro de lo normal (hallazgo que se utiliza para realizar el diagnóstico de FQ) y con una proporción entre cloro negativo y sodio positivo alterada. Esto hace que los pacientes con FQ tengan unas pérdidas hidroelectrolíticas aumentadas por esta vía, lo que les somete a un riesgo especial en situaciones ambientales extremas (calor o ejercicio físico intenso) y en épocas de la vida con más riesgo de deshidratación, como la lactancia.
Una forma de presentación, que también puede manifestarse como complicación en el paciente ya diagnosticado, es la deshidratación moderada o grave con alcalosis metabólica hipoclorémica e hipopotasémica, en ocasiones ante procesos intercurrentes que parecen no justificar la intensidad del cuadro o, incluso, sin factor precipitante alguno evidente.
Síndrome de anemia-edemashipoproteinemiaEsta tríada clínica puede presentarse en lactantes con alteraciones mucosas intestinales difusas o de la función pancreática que condicionen una malabsorción con malnutrición y edemas hipoproteinémicos. Aunque no es exclusiva de la FQ su presencia obliga a descartarla.
Colestasis neonatalAlrededor de un 5% de los recién nacidos afectados de FQ pueden presentar cuadros de ictericia prolongada. A veces, estos cuadros pueden ser intensos y persistentes, e incluso plantean el diagnóstico diferencial con la atresia biliar. Por ello, es imperativo descartar la existencia de una FQ en los lactantes con colestasis prolongada, especialmente si hay el antecedente de un íleo meconial9.
Este cuadro suele resolverse espontáneamente en los primeros meses de vida, y no necesariamente progresa hacia otras formas de hepatopatía asociada a la FQ.
Otras manifestaciones digestivasAfectación hepáticaComo ya hemos comentado, el CFTR se expresa en la superficie de los colangiocitos y de las células epiteliales de la vesícula biliar, dando lugar a manifestaciones clínicas en un tercio de los pacientes9,10.
Es una complicación más frecuente hacia el final de la primera década de la vida, siendo raro su inicio posterior, excepto en pacientes con antecedente de íleo meconial, factor de riesgo para el desarrollo de hepatopatía.
La afectación suele comenzar con una fibrosis periportal que evoluciona de forma lenta y progresiva, en la que predominan los síntomas de hipertensión portal frente a los de insuficiencia hepatocelular que, cuando se presentan, son mucho más tardíos. Estos pacientes tienen más riesgo de presentar, además, otras manifestaciones clínicas de mal pronóstico, y se ha descrito más incidencia de malnutrición y osteoporosis en los pacientes con FQ con afectación hepática.
Las alteraciones bioquímicas son leves o intermitentes, y no guardan relación con los hallazgos histológicos encontrados cuando se realiza biopsia hepática, que pueden oscilar desde una esteatosis (generalmente secundaria a malnutrición) o una fibrosis periportal leve, hasta una cirrosis biliar multinodular.
Otras manifestaciones hepatobiliares frecuentes son la vesícula escleroatrófica, presente en cerca del 30% de los pacientes, y la colelitiasis, en alrededor del 15%.
Síndrome de obstrucción intestinalSe presenta en el 15% de los pacientes con FQ y es casi exclusiva de los pacientes con insuficiencia pancreática. se denominaba anteriormente equivalente del íleo meconial, debido al parecido en su mecanismo de producción, basado en la obstrucción, parcial o total, del intestino delgado distal por la presencia de contenido intestinal con mayor viscosidad, lo que dificulta el tránsito digestivo normal. Es una complicación típica de escolares y adolescentes, pero pueden observarse también cuadros similares en niños más jóvenes con antecedente de íleo meconial, especialmente si coexisten secuelas de una cirugía neonatal.
La presentación suele ser insidiosa, con dolor abdominal recurrente en fosa ilíaca derecha, masa palpable a ese nivel y evidencia radiológica de retención de heces en la zona del íleon terminal y colon derecho. En ocasiones, ya sea desde el inicio o tras la evolución descrita, la obstrucción puede ser total, y entonces se produce un cuadro clínico de obstrucción intestinal indistinguible del producido por cualquier otra causa mecánica.
Una vez producido, el tratamiento se basa en el uso de soluciones de lavado con polietilenglicol, la corrección de las alteraciones hidroelectrolíticas que pueda haber asociadas y la regulación de la dosis de enzimas pancreáticas una vez reiniciada la alimentación oral. sin embargo, en casos resistentes al tratamiento o en pacientes en situación crítica, puede ser necesario el tratamiento quirúrgico.
La prevención se basará en optimizar las dosis de suplementos pancreáticos (tanto el exceso, como el defecto, como los cambios bruscos e intensos de dosificación pueden favorecer esta complicación) y regularizar el hábito intestinal, valorando la utilización de acetilcisteína o dosis bajas de soluciones de lavado de mantenimiento en niños con episodios recidivantes.
Prolapso rectalSe produce en un 20% de los pacientes, siendo en la mitad de ellos el síntoma de inicio de la enfermedad. Es más frecuente en menores de 5 años y su aparición está producida por una serie de factores, como la presencia de heces voluminosas por la insuficiencia pancreática, la disminución del trofismo del suelo de la pelvis por la malnutrición y, en algunos casos, el aumento de la presión intraabdominal debida a la tos o a maniobras de fisioterapia respiratoria11. El prolapso suele ser mucoso, por lo que únicamente está indicada su reducción manual y la corrección de los factores predisponentes. sin embargo, en una pequeña proporción, los episodios pueden persistir, ser de reducción dificultosa o dolorosa o asociarse con incontinencia. sólo en estos casos estaría indicado valorar el tratamiento quirúrgico.
Reflujo gastroesofágicoUn 20-25% de los pacientes experimenta pirosis y/o sensación de regurgitación, especialmente adolescentes o preadolescentes con afectación respiratoria importante, en los cuales el reflujo se favorece por la propia afectación pulmonar y por las maniobras de fisioterapia. De este modo, el riesgo de esofagitis es mayor en pacientes con neumopatía grave.
El tratamiento antirreflujo debe ser temprano e intenso. Las indicaciones de la cirugía antirreflujo son controvertidas, ya que estos pacientes presentan un porcentaje más elevado de fracasos de la técnica quirúrgica, por lo que hay autores que proponen el tratamiento con inhibidores de la bomba de protones de manera continua en los casos de reflujo gastroesofágico con esofagitis12.
Intolerancia a los hidratos de carbonoAunque inicialmente la lesión pancreática afecta su función exocrina, evolutivamente puede producirse también una disminución de islotes de Langerhans, de modo que hasta un 40% de los pacientes llega a presentar una intolerancia a los hidratos de carbono por encima de los 10 años de vida, y alrededor de un 10% desarrolla una diabetes franca que precisa tratamiento hipoglucemiante13,14.
La intolerancia puede detectarse durante un control sistemático o ponerse de manifiesto mediante un deterioro clínico general, especialmente en las situaciones de más estrés metabólico, como son las agudizaciones respiratorias.
Se distinguen 5 situaciones en función de los datos analíticos de glucemia basal en ayunas y de sobrecarga con glucosa15,16:
- 1.
Tolerancia normal a la glucosa: en ayunas inferior a 110mg/dl y a las 2h de la sobrecarga inferior a 140mg/dl.
- 2.
Alteración de la glucemia en ayunas: entre 110 y 126mg/dl en ayunas, inferior a 140mg/dl a las 2h de la sobrecarga.
- 3.
Alteración de la tolerancia a la glucosa: inferior a 126mg/dl en ayunas, con glucemias entre 140 y 199mg/dl a las 2h de la sobrecarga.
- 4.
Diabetes relacionada con la FQ (DRFQ) sin hiperglucemia en ayunas: inferior a 126mg/dl en ayunas y superior a 200mg/dl a las 2h de la sobrecarga.
- 5.
DRFQ con hiperglucemia en ayunas: superior a 126mg/dl en ayunas y a 200mg/dl a las 2h de la sobrecarga.
La DRFQ comparte muchos aspectos con la diabetes mellitus dependiente de la insulina, pero guarda también algunos aspectos diferenciadores. Por ejemplo, resulta muy infrecuente la producción de cetoacidosis, ya que la destrucción del islote es global y hay un déficit de hormonas contrarreguladoras, al contrario de la diabetes mellitus, en la que se afectan selectivamente las células beta. Estas diferencias condicionan algunos aspectos de su tratamiento clínico, que se tratarán más adelante.
Colonopatía fibrosanteEntre los años 1993 y 1996, se describieron más de 80 casos de esta rara complicación de la FQ17,18. Este cuadro se caracteriza por la aparición de forma insidiosa de síntomas similares a los del soID que progresan hasta culminar en un cuadro de obstrucción intestinal con síntomas de colitis. Los estudios de imagen mostraban una obstrucción localizada en la zona del colon derecho, con pérdida de haustración. Los datos histológicos, característicos de esta entidad, consistían en un engrosamiento extenso submucoso debido a la presencia de una banda de colágeno que respeta la submucosa, con un epitelio colónico íntegro. El tratamiento consistía en la resección quirúrgica del segmento de colon afectado.
La colonopatía fibrosante se relacionó con el uso de suplementos pancreáticos a altas dosis y con cubierta entérica que contenía un copolímero de ácido metacrílico (minitabletas con Eudragit). una vez retirados estos preparados y evitado el uso de dosis excesivamente elevadas de enzimas, esta complicación prácticamente ha desaparecido, y únicamente se han comunicado casos esporádicos.
Lectura rápida
Las pruebas más útiles para el diagnóstico de la insuficiencia pancreática se basan en la medición de enzimas fecales, especialmente quimotripsina y elastasa.
La afectación hepática se presenta en una cuarta parte de los pacientes, y hay una correlación pobre entre los hallazgos histológicos y las manifestaciones clínicas.
Quizá la alteración más utilizada con fines diagnósticos sea la determinación de electrolitos en sudor. Además, hay otras técnicas diagnósticas encaminadas a valorar la presencia de insuficiencia pancreática o de afectación hepática, además de las pruebas de control y seguimiento desde el punto de vista respiratorio, que no trataremos en este trabajo.
Determinación de electrolitos en sudorLa producción de un sudor con mayor contenido de cloro negativo y sodio positivo se utiliza con fines diagnósticos.
La prueba consiste en realizar una iontoforesis inducida por pilocarpina y una corriente eléctrica de baja intensidad, recogiéndose el sudor para su posterior análisis19. El ionotest es el método más conocido y determina la concentración de cloro negativo, considerándose patológica por encima de 60mEq/l, normal por debajo de 40mEq/l y dudosos los resultados intermedios. No obstante, el ionotest se considera un método de cribado, por lo que su resultado debe confirmarse midiendo la conductividad de cloro negativo, que se considera patológica cuando es mayor de 50mEq/l.
Otros métodos, como la cristalización del sudor o la determinación del conciente cloro:sodio, son mucho menos fiables y no sirven para confirmar el diagnóstico de la enfermedad.
Estudio de función pancreáticaLos métodos directos, como el sondaje duodenal con estimulación con colecistocinina-secretina, son los que tienen más sensibilidad y especificidad, pero rara vez se utilizan en clínica debido a su complejidad e invasividad. Habitualmente, se utilizan métodos indirectos, como la determinación de grasa en heces o la concentración de enzimas pancreáticas en sangre (amilasa, lipasa, tripsina sérica) o en heces (quimotripsina y elastasa)20.
El estudio de la eliminación de grasa se realiza habitualmente mediante la recogida de heces de 3 días y su posterior análisis, ya sea por el método de Van de Kamer o mediante la absorción en el infrarrojo cercano (FENIR). Puede valorarse la excreción fecal de grasa (normal hasta 3–4g/24h en niños y hasta 6g/24h en adolescentes y adultos), aunque lo más correcto es calcular el cociente de absorción de grasas, para lo cual es preciso combinar la prueba con una encuesta dietética y una calibración de la dieta ingerida (valor normal: 80-85% en lactantes, 85-95% en escolares y > 95% en adolescentes). otros métodos, como la determinación del esteatocrito fecal, aunque más sencillos de realizar, son menos fiables. Por último, hay que tener en cuenta que hay otros factores que contribuyen a la presencia de esteatorrea: alteraciones de motilidad, hiperacidez duodenal secundaria a hipersecreción gástrica (y menor secreción pancreática de bicarbonato), pérdida de sales biliares e inactivación de los suplementos pancreáticos por la hiperacidez.
La determinación de enzimas séricas tiene el inconveniente de su menor sensibilidad y de no informar del grado de función residual del páncreas exocrino. La determinación de tripsina sérica es la base del cribado neonatal de la FQ ya que se encuentra elevada en recién nacidos y lactantes con insuficiencia pancreática. su valor cae rápidamente desde las primeras semanas de vida, y alcanza valores mínimos por encima de los 7 años de edad. Por tanto, fuera de los primeros meses de vida no es útil para diferenciar entre suficientes e insuficientes pancreáticos entre la lactancia y los 7 años.
Más útil en este caso sería la determinación de enzimas en heces. La quimotripsina tiene el inconveniente de afectarse por el tratamiento sustitutivo (aunque no sirve para ajustar la dosis de los suplementos), por lo que en la actualidad tiende a utilizarse la determinación de elastasa, con mayor sensibilidad y especificidad para detectar la insuficiencia pancreática y que puede determinarse de forma seriada sin necesidad de suspender el tratamiento con enzimas. Las pruebas de imagen (habitualmente ecografías seriadas) muestran los cambios habituales en los pacientes con insuficiencia pancreática: aumento de la hiperecogenicidad pancreática debida a la fibrosis progresiva, con disminución del tamaño de la glándula. Pueden observarse también formaciones quísticas pancreáticas, en ocasiones incluso de gran tamaño21. Estos métodos, sin embargo, no informan acerca de la función pancreática residual.
Estudio de la afectación hepáticaLa afectación hepática es, con frecuencia, subclínica, y únicamente es evidente cuando el daño ya es difuso e importante. La combinación de exploración física (hepatomegalia) y determinación de enzimología hepática se realiza de forma seriada, pero puede fracasar en detectar la hepatopatía, además de no guardar correlación con su intensidad9.
La ecografía es más útil para valorar el estado de la vesícula biliar y el tamaño del hígado y sirve para medir los flujos arteriales hepáticos, pero puede no detectar los cambios iniciales. El hallazgo más frecuente de forma temprana suele ser la esteatosis, seguida posteriormente de signos de fibrosis periportal (hallazgo más característico) con la posible evolución posterior hacia un hígado cirrótico. Puede también demostrar la existencia de una hipertensión portal o de alteraciones de la vía biliar. Otras pruebas de imagen, como la tomografía computarizada, la resonancia magnética o la colangiografía, no aportan grandes ventajas y tienen el inconveniente de ser más caras, precisar sedación en niños y, en el caso de la colangiografía, estar sujetas a complicaciones. La gammagrafía informa sobre las alteraciones del drenaje biliar y sobre alteraciones en vías biliares, por lo que se ha utilizado en ocasiones para controlar el tratamiento médico.
La biopsia hepática detecta alteraciones tempranas, pero tiene el inconveniente de su invasividad y de que puede dar resultados no representativos por error de obtención, ya que la lesión hepática puede ser parcheada. En cualquier caso, la biopsia proporciona información importante acerca del tipo predominante de lesión (esteatosis o cirrosis biliar focal), del grado de fibrosis portal y de la velocidad de progresión tras iniciar el tratamiento. Por tanto, se indica cuando hay dudas acerca del diagnóstico, para establecer el diagnóstico de cirrosis y previa al tratamiento médico o al trasplante hepático9.
Estudio genéticoEl estudio genético puede ser suficiente para establecer el diagnóstico de FQ, siempre que se detecten 2 mutaciones claramente relacionadas con la enfermedad. Esto es especialmente útil en casos de diagnóstico dudoso o de clínica atípica, como puede ser el caso de varones con escasa sintomatología digestiva o respiratoria y presencia de azoospermia obstructiva por agenesia bilateral de vasos deferentes.
Además, el estudio genético familiar permite realizar consejo genético y, mediante la biopsia corial durante el embarazo, detectar la afectación del embrión para poder realizar la interrupción de la gestación, si así se desea.
Cribado neonatalDurante tiempo se ha debatido la utilidad de realizar pruebas de cribado neonatal en toda la población para la detección temprana de la enfermedad. Estas pruebas suelen realizarse en sangre de talón y se basan en la determinación de tripsina sérica.
Parece que los lactantes que comienzan seguimiento antes de iniciar los síntomas podrían tener mejor pronóstico y menor riesgo de complicaciones, con menores costes derivados del tratamiento que los de niños diagnosticados clínicamente. Por tanto, los estudios realizados recientemente concluyen que los beneficios clínicos, sociales y económicos aconsejarían realizar el cribado neonatal de FQ de forma universal22.
Lectura rápida
El diagnóstico se basa en la demostración del CFTR, habitualmente mediante el estudio de electrolitos en sudor, junto con clínica compatible y/o familiaridad de primer grado para la enfermedad.
De manera alternativa, el hallazgo de 2 mutaciones reconocidas basta para realizar el diagnóstico de fibrosis quística.
Como ya hemos comentado, el hallazgo de 2 mutaciones claramente relacionadas con la enfermedad es suficiente para realizar el diagnóstico de FQ.
De otra forma, se requiere la demostración de la presencia del CFTR asociado a un fenotipo compatible de FQ o a familiaridad de primer grado para la enfermedad23.
La forma más habitual de demostrar la presencia de un CFTR anómalo es mediante la determinación del cloro negativo en sudor en 2 ocasiones. otros métodos son la demostración de la insuficiencia pancreática con disminución de la secreción de bicarbonato tras la estimulación y la determinación de la diferencia de potencial nasocutáneo, aunque apenas se emplean en clínica.
Se considera fenotipo compatible con la enfermedad la presencia de uno o varios de los siguientes:
- —
Enfermedad pulmonar crónica.
- —
Alteraciones intestinales: íleo meconial, insuficiencia pancreática exocrina, SOID, prolapso rectal, pancreatitis recurrente.
- —
Enfermedad hepatobiliar crónica: cirrosis biliar focal o multinodular.
- —
Fracaso del desarrollo con malnutrición calórico-proteica.
- —
síndrome de anemia-edemas-hipoproteinemia.
- —
Déficit de vitaminas liposolubles.
- —
Azoospermia obstructiva.
- —
Síndrome de pérdida hidrosalina.
Desde el punto de vista digestivo, el tratamiento general de la FQ se divide en 3 aspectos fundamentales: tratamiento sustitutivo de la insuficiencia pancreática, tratamiento de la hepatopatía en aquellos que la presenten y soporte nutricional adecuado para cubrir las necesidades aumentadas y evitar las complicaciones derivadas del fracaso nutricional.
Insuficiencia pancreáticaSe basa en la administración de enzimas pancreáticas, compuestas por microesferas que contienen lipasa y proteasa protegidas por una cubierta entérica resistente al pH ácido del estómago. Estas enzimas deben tomarse antes de las comidas, y evitar su administración con bebidas alcalinas o, en caso de abrir la cápsula y dar directamente las microesferas, evitar masticarlas, ya que en ambas situaciones podría destruirse la cubierta e inactivarse el preparado en el estómago.
La dosis debe individualizarse en cada paciente. Hay cierto acuerdo en torno a la dosis inicial (tabla 1), que posteriormente se ajusta en función de la ingesta dietética (fundamentalmente de grasa), el estado nutricional y las características de las deposiciones24.
Hay que tener en cuenta que la absorción de grasa depende de otros factores diferentes de la función pancreática, por lo que no siempre se consigue normalizar la eliminación fecal a pesar de aumentar la dosis de enzimas. Cuando se alcanzan las 1.200 unidades de lipasa/kg de peso/comida es conveniente revisar si se toman de forma regular y adecuada, y valorar otras medidas (como el tratamiento antisecretor para disminuir la acidez duodenal, que puede interferir con la actividad de los suplementos pancreáticos) antes de aumentar progresivamente la dosis hasta valores demasiado elevados.
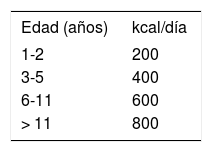
Como ya se ha comentado, la esteatorrea puede producir un déficit de vitaminas liposolubles, por lo que estos pacientes tienen unas necesidades que doblan las de los niños de su edad y sexo (tabla 2). Además, todos estos pacientes reciben también suplemento con vitaminas hidrosolubles, generalmente con uno de los preparados polivitamínicos de uso habitual.
Dosificación inicial de los suplementos con vitaminas liposolubles en pacientes con fibrosis quística
| Vitaminas | Suplemento |
|---|---|
| Vitamina A (U/día) | 5.000-10.000 |
| Vitamina D (U/día) | 400-800 |
Vitamina E (U/día)
|
|
Vitamina K (mg/semana)
|
|
El riesgo de malnutrición es alto en pacientes con FQ en relación con factores diversos, como la disminución de la ingesta, la malabsorción y el aumento del gasto producido fundamentalmente por el cuadro respiratorio crónico. Por todo ello, estos pacientes necesitan una dieta que aporte los elevados requerimientos de nutrientes que precisan25.
- A.
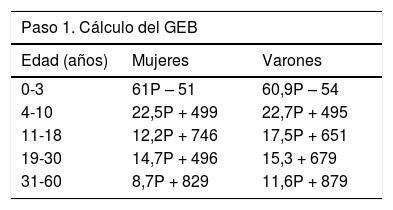
Energía. Como norma general, se recomiendan unos aportes energéticos de entre el 120 y el 130% de las RDA (del inglés recommended dietary allowances) para edad y sexo, aunque puede ser necesario aumentar esta cantidad en casos de fracaso nutricional. Cuando no sea posible calcular el gasto mediante calorimetría indirecta, pueden utilizarse la fórmulas propuestas por el Comité de Consenso de la Cystic Fibrosis Foundation (tabla 3)26.
Tabla 3.Cálculo de los requerimientos energéticos diarios en pacientes con fibrosis quística según las recomendaciones del Comité de Consenso de la Cystic Fibrosis Foundation
Paso 1. Cálculo del GEB Edad (años) Mujeres Varones 0-3 61P – 51 60,9P – 54 4-10 22,5P + 499 22,7P + 495 11-18 12,2P + 746 17,5P + 651 19-30 14,7P + 496 15,3 + 679 31-60 8,7P + 829 11,6P + 879 Paso 2. Calcular el GET, multiplicando el GEB por el CA y sumando el factor de corrección, según la enfermedad A. Coeficientes de actividad enfermedad
Encamado: GEB x 1,3
Sedentario: GEB x 1,5
Activo: GEB x 1,7B. Factores de corrección según
FEV1 > 80%: GEB x (CA + 0)
FEV1 40-80%: GEB x (CA + 0,2)
FEV1 < 40%: GEB x (CA + 0,3) hasta 0,5 en casos de neumopatía muy gravePaso 3. Calcular los requerimientos diarios teniendo en cuenta el grado de esteatorrea Requerimientos = GET x (0,93/coeficiente de absorción de grasa) CA: coeficiente de actividad; FEV1: volumen espiratorio máximo en el primer segundo; GEB: gasto energético basal;
GET: gasto energético total; P: peso en kg.
- B.
Proteínas. Deben aportar un 12-15% de volumen calórico total (VCT) (200% de las RDA), y hay que prestar atención a que al menos dos terceras partes sea proteína de alto valor biológico. En lactantes, no se contraindica la lactancia materna ni es obligatorio administrar una fórmula hidrolizada, si el paciente no está gravemente malnutrido27, aunque sí parece aconsejable administrar suplemento con enzimas a aquéllos que tengan insuficiencia pancreática.
- C.
Hidratos de carbono. Deben aportar entre el 35 y el 45% del VCT. si hay alteración de tolerancia a la glucosa o diabetes, deberá evitarse el consumo excesivo de hidratos de carbono complejos, así como de bebidas azucaradas, y fomentar la ingesta de hidratos de carbono no refinados. Los suplementos nutricionales, si se emplean, es preferible que no sean sacarosados, especialmente en los pacientes que no reciben tratamiento insulínico28. En los pacientes con DRFQ debe prestarse atención a no hacer restricción calórica para conseguir una regulación mejor de la glucemia, y en su lugar administrar medicación hipoglucemiante, si fuese necesario.
- D.
Lípidos. Aportarán entre el 35 y el 45% del VCT, prestando atención al consumo de ácidos grasos esenciales (1-3% del VCT en forma de linoleico). En la FQ hay una alteración del equilibrio de ácidos grasos esenciales, con una disminución del cociente araquidónico/docosaexanoico que puede influir en la patogenia de la enfermedad y en la producción de algunas de sus manifestaciones29,30.
- E.
Minerales y oligoelementos. Es importante prestar atención a los valores de cinc y hierro, suplementándolos si es necesario31. Además, es conveniente administrar suplementos de cloruro sódico a los pacientes menores de 2 años, a los que presenten manifestaciones clínicas de pérdida hidrosalina y a todos, en general, en condiciones de calor o ejercicio físico intenso. Debe realizarse un seguimiento nutricional en todos los pacientes, y procurar que la dieta cumpla las características comentadas. En los casos en los que el desarrollo nutricional no sea óptimo, se deberá establecer un soporte nutricional más intervencionista, que abarque desde la modificación de los hábitos dietéticos habituales, hasta la administración de suplementos nutricionales por vía oral o parenteral32,33.
Como suplementos suelen emplearse dietas poliméricas completas, generalmente hipercalóricas e hiperproteicas, aunque pueden emplearse también los polímeros de glucosa (siempre que no haya alteraciones del metabolismo de los hidratos de carbono) (tabla 4)26.
Cuando persiste un fracaso nutricional a pesar de estas medidas, se instaura una nutrición enteral a débito continuo, generalmente de tipo nocturno y a través de gastrostomía, que aporte alrededor del 40-50% de los requerimientos diarios. La utilización de nutrición parenteral queda reservada para situaciones de intestino corto y poscirugía, cuando se rechace la nutrición enteral y haya un fracaso nutricional importante y, ocasionalmente, para la rehabilitación nutricional previa a trasplante hepático o pulmonar.
Tratamiento de la hepatopatíaEl tratamiento de la afección hepática en la FQ ha cobrado más importancia al aumentar la supervivencia de estos pacientes. El fármaco que se emplea habitualmente es el ácido ursodeoxicólico, que tiene efectos beneficiosos porque reemplaza los ácidos biliares hidrofóbicos retenidos en los cuadros colestáticos, además de tener un efecto citoprotector directo por estimular la secreción de cloro negativo a través de canales dependientes de Ca2+. Se emplea a dosis más elevadas respecto a otras situaciones de colestasis (20mg/kg/día) debido a la disminución del conjunto de sales biliares presente en la FQ. Sin embargo, aunque está claro que mejora la función excretora hepática y las alteraciones analíticas, está por aclarar su verdadero impacto en la evolución natural de la enfermedad9.
Cuando hay manifestaciones debidas a la hipertensión portal grave, se emplean las medidas terapéuticas habituales para la hipertensión portal, y que escapan al ámbito de esta revisión.
Por último, el trasplante hepático estaría indicado en pacientes con insuficiencia hepática y/o manifestaciones de hipertensión portal que pongan en peligro la vida del paciente, siempre que el grado de afectación pulmonar permita esperar una supervivencia prolongada del paciente.
Pronóstico y desarrollos futurosCon la mejora progresiva de los medios terapéuticos y de soporte nutricional, junto al tratamiento de estos pacientes en unidades especializadas, se ha conseguido aumentar la calidad de vida y la supervivencia de esta enfermedad. En la actualidad, la mayoría de los pacientes alcanzan la vida adulta, con una esperanza de vida en torno a los 40 años6.
Hay una serie de procesos y enfoques terapéuticos en desarrollo encaminados a disminuir aún más la morbilidad y la mortalidad de estos pacientes.
Desde el punto de vista del tratamiento sustitutivo, se trabaja en la producción de suplementos pancreáticos más potentes, con una cubierta entérica más eficaz y con microesferas de menor tamaño, con lo que se disminuirá el número de cápsulas que deben tomar muchos pacientes y se facilitará su administración en lactantes34. Se desarrollan también nuevos preparados hidrosolubles de vitaminas liposolubles, con lo que aumentará la biodisponibilidad del fármaco35.
Por otra parte, se desarrollan continuamente nuevos productos nutricionales adaptados a la situación fisiopatológica de la FQ, tanto en forma de fórmulas hidrolizadas, como de fórmulas poliméricas completas y suplementos. Además, se refinan las técnicas de nutrición enteral, con mejoras en el ámbito de bombas, sondas de gastrostomía, etc.
Queda pendiente aclarar el verdadero papel que desempeña el desequilibrio de ácidos grasos esenciales en la membrana celular, ya que podría abrirse una nueva vía terapéutica basada en el suplemento de ácidos grasos de serie omega-3, como el docosaexanoico, aunque estudios recientes ponen en duda la utilidad de este tratamiento y cuestionan el hecho de que la alteración de ácidos grasos sea secundaria al cuadro general, y no causa primaria36.
Hay grandes expectativas en los resultados que puedan producirse con el tratamiento génico, con la incorporación de genes que permitan la síntesis de un CFTR funcionante, pero estos tratamientos se encuentran aún lejos de su uso en clínica debido a la poca eficacia de transferencia génica conseguida hasta la actualidad y a lo transitorio de los efectos logrados.
Por último, quizá lo más novedoso sean las modalidades de tratamiento molecular, diseñadas específicamente según el tipo de mutación que porte el paciente6. En este sentido, parece que algunos fármacos, como la gentamicina, permiten leer a través de las señales de parada de algunas mutaciones de clase I.
Otros, como el fenilbutirato o el s-nitrosoglutatión, pueden inhibir la función de las chaperonas y permitir que el CFTR alcance la membrana en determinadas mutaciones de clase ii. otras sustancias, como la genisteína o la milrinona, consiguen aumentar la funcionalidad del CFTR en ciertas mutaciones de clases III y IV. Por último, hay numerosas sustancias en fase de ensayo para aumentar el rendimiento de los otros transportadores de iones (Moli-1901, INS37217, SPI-8611, Vertex), con lo que podría paliarse la alteración de la interfase hidroelectrolítica de los epitelios afectados.
De los resultados de todos estos ensayos en curso surgirán, sin duda, nuevos fármacos que permitirán mejorar día a día tanto la calidad de vida, como la supervivencia de los pacientes con FQ.