














La fibrosis quística (FQ) es la enfermedad hereditaria, autosómica, recesiva, grave, más frecuente en la raza blanca, con una gran variabilidad en la incidencia entre países y razas. La mayoría de los autores estiman una incidencia entre 1/2.500 y 1/5.000 recién nacidos vivos1. En España, a raíz de los programas de detección neonatal implantados en algunas comunidades a partir del año 1999, se estima que la incidencia está en torno a 1/1.450 en Castilla y León2 y 1/5.240 en Cataluña3.
La enfermedad se produce como consecuencia de la alteración del gen CFTR (del inglés cystic fibrosis transmembrane conductance regulator), situado en el brazo largo del cromosoma 74. Este gen codifica la proteína CFTR, que regula el transporte iónico, por lo que su alteración determina un transporte anormal en las células epiteliales de diferentes sistemas y órganos, principalmente en los tractos gastrointestinal y respiratorio.
La afectación pulmonar y sus complicaciones son las que condicionan la mayor morbimortalidad, ya que serán la causa del 95% de los fallecimientos en estos pacientes.
En el ámbito pulmonar, la proteína CFTR anómala, situada en la membrana apical de las células del epitelio bronquial hace que en las células epiteliales, en lugar de haber un transporte activo de cloro desde el intersticio hacia la luz y de reabsorción de sodio en dirección opuesta, suceda lo contrario, produciendo una dificultad en la excreción de cloro y una mayor reabsorción de sodio, que da lugar a una hiperviscosidad de moco que deteriora el aclaramiento mucociliar. Se produce obstrucción bronquial y una respuesta inflamatoria anormal con susceptibilidad a la infección endobronquial por bacterias específicas.
Curiosamente, los pulmones del recién nacido son histológicamente normales. Sin embargo, ya en los primeros meses de la vida, algunos pacientes desarrollan una colonización bacteriana crónica endobronquial. En la fase inicial es característica la presencia de Haemophilus influenzae y/o Staphylococcus aureus. Posteriormente, la casi totalidad de los pacientes presenta colonización por Pseudomonas aeruginosa, que se asocia a un deterioro progresivo de la función pulmonar.
No obstante, en los últimos años la supervivencia se ha incrementado claramente debido a la creación de unidades de FQ y a los nuevos tratamientos, de forma que según los datos de la Fundación Americana de Fibrosis Quística, en 2006, la mediana de la expectativa de vida llegaba hasta los 37 años5.
Puntos clave

La edad de comienzo de los síntomas respiratorios es muy variable. A diferencia de los síntomas digestivos y la deshidratación, que pueden aparecer de forma más temprana, la clínica respiratoria puede aparecer en la primera infancia o, en formas más leves, en la adolescencia o en adultos jóvenes. En el lactante, puede iniciarse con clínica de tos persistente, clínica de bronquiolitis, a veces fuera de una época epidémica, o tos pertusoide. Progresivamente, según evoluciona la enfermedad, se desarrollan bronquiectasias y aparece tos productiva. Algunos pacientes cursan con hiperreactividad bronquial y son diagnosticados de asma.
SíntomasLa tos es uno de los primeros síntomas; persistente, inicialmente seca y después productiva, con mala respuesta a los tratamientos.
Lectura rápida
La fibrosis quística (FQ) es la enfermedad hereditaria autosómica recesiva grave más frecuente en la raza blanca. En España, a raíz de los programas de cribado neonatal implantados en algunas comunidades a partir del año 1999, se estima que la incidencia está en torno a 1/4.000–5.000 nacidos vivos.
FisiopatologíaLa enfermedad se produce como consecuencia de la alteración del gen CFTR (del inglés cystic fibrosis transmembrane conductance regulator) situado en el brazo largo del cromosoma 7. Este gen codifica la proteína CFTR que regula el transporte iónico intracelular, por lo que su alteración determina un transporte anormal en las células epiteliales de diferentes sistemas y órganos, principalmente en los tractos gastrointestinal y respiratorio.
En el ámbito pulmonar, la proteína CFTR anómala, situada en la membrana apical de las células del epitelio bronquial, hace que en las células epiteliales, en lugar de haber un transporte activo de cloro desde el intersticio hacia la luz y de reabsorción de sodio en dirección opuesta, suceda lo contrario, produciendo una dificultad en la excreción de cloro y una reabsorción mayor de sodio, que da lugar a una hiperviscosidad de moco que deteriora el aclaramiento mucociliar. Se produce obstrucción bronquial y una respuesta inflamatoria anormal con susceptibilidad a la infección endobronquial por bacterias específicas.
El segundo síntoma de importancia es la expectoración, con cambios en la cantidad, la viscosidad y el color, en relación con las reagudizaciones y la colonización bacteriana.
La dificultad respiratoria puede confundirse en las primeras etapas con síntomas de una bronquiolitis o de asma, pero se diferencia por su cronicidad, recurrencia y por la escasa respuesta a los tratamientos broncodilatadores.
La sinusitis es constante en estos pacientes. La presencia de pólipos nasales en un niño obliga siempre a descartar esta enfermedad.
ExploraciónLos signos clínicos respiratorios varían desde una exploración prácticamente normal a cualquiera de los signos que se indican a continuación y que guardan relación con la gravedad de la enfermedad:
- —
Deformidad torácica (cifosis y aumento del diámetro anteroposterior del tórax) secundaria a hiperinsuflación pulmonar.
- —
Aumento de la frecuencia respiratoria con tiraje subcostal, intercostal y supraclavicular.
- —
Alteración de la auscultación pulmonar (estertores, sibilancias).
- —
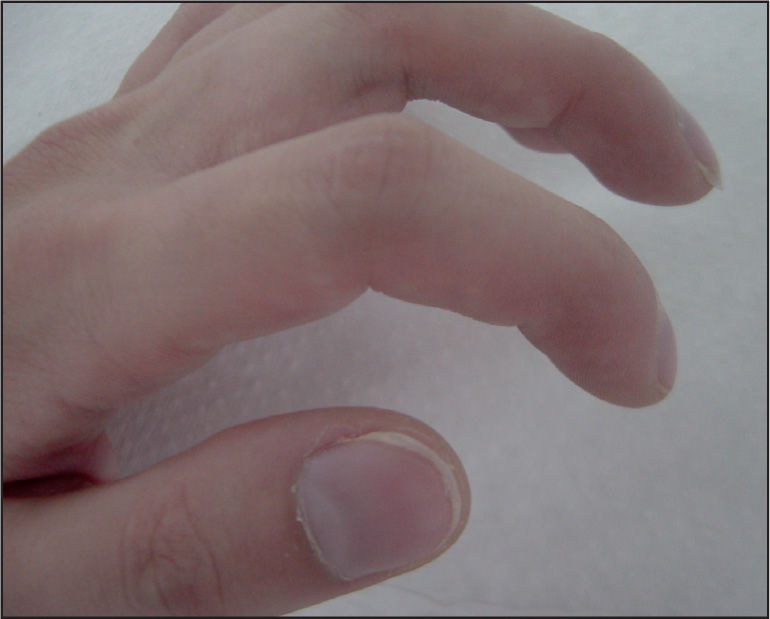
Acropaquias (su existencia obliga a excluir una FQ) (fig. 1).
- —
Pólipos nasales.

En los primeros años puede ser normal, pero, según avanza la enfermedad, presentan en general un patrón obstructivo con afectación del volumen espiratorio máximo en el primer segundo y, sobre todo, de los mesoflujos. Por último se observa un patrón mixto obstructivo y restrictivo con atrapamiento aéreo.
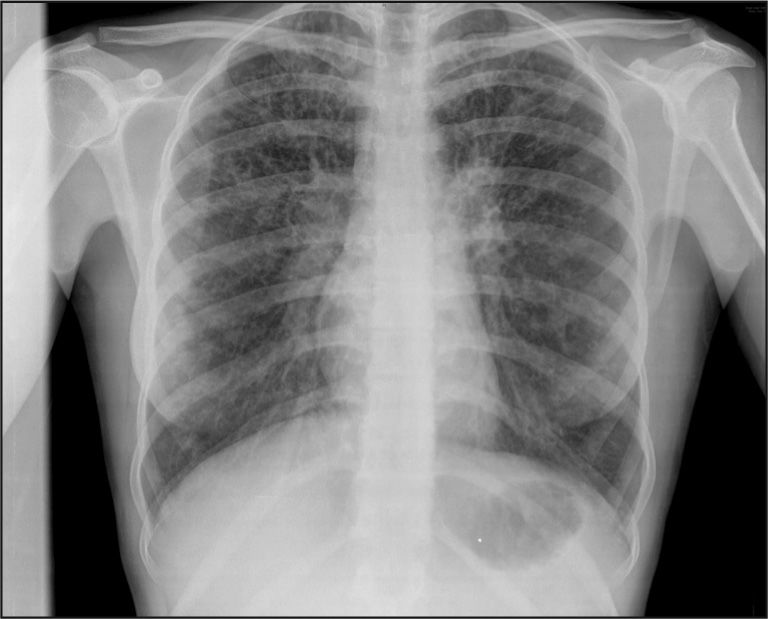
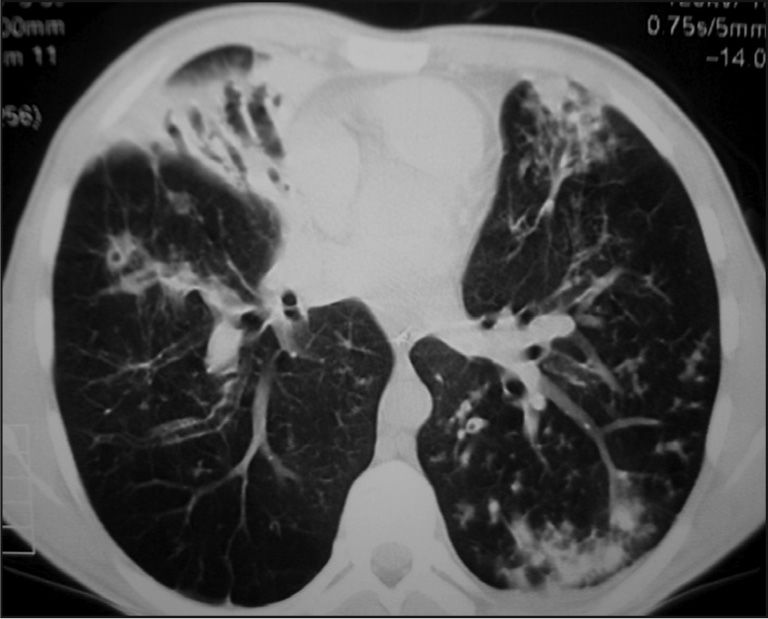
RadiologíaComo ocurre con la clínica, puede ser muy variable. Desde una radiología prácticamente normal en formas leves o en los primeros años, hasta las formas clásicas con hiperinsuflación pulmonar progresiva, inflamación bronquial, bronquiectasias claras, afectación alvéolo-intersticial, condensaciones, atelectasias, etc.
De forma sistemática, se realizan radiografías de tórax y tomografías computarizadas (TC), según las pautas de los distintos centros de FQ y las necesidades individuales6 (figs. 2 y 3).


En el ámbito sinusal, sinusitis y mucoceles.
Evolución respiratoriaEs muy variable en función de diversos factores genéticos, ambientales, sociales, etc. Durante la evolución, presentan procesos intercurrentes con exacerbaciones respiratorias.
Las exacerbaciones respiratorias se diagnostican por el incremento de la tos, cambios en el esputo, inicio o aumento de la disnea, incremento en la frecuencia respiratoria, empeoramiento de la auscultación pulmonar, cambios radiológicos, empeoramiento de la función pulmonar en niños colaboradores y peor tolerancia al ejercicio. Excepcionalmente puede asociarse fiebre y leucocitosis.
Las exacerbaciones respiratorias se deben a diversos patógenos:
- 1.
Bacterias: en los estadios iniciales H. influenzae y/o S. aureus desplazados por P aeruginosa posteriormente. A partir de los 10 años, aumentan su incidencia otros patógenos oportunistas multirresistentes (Burkholderia cepacia, Alcoligenes xylosoxidans, Stenotrophomonas maltophilia).
- 2.
Virus: causantes de alrededor de un 40% de las exacerbaciones, que conducen a un deterioro que secundariamente predispone a la infección/colonización bacteriana.
- 3.
Hongos: los más frecuentes aislados son Candida albicans y Aspergillus fumigatus.
- 4.
Micobacterias no tuberculosas: ha aumentado la frecuencia de su aislamiento en las secreciones, pero se desconoce aún su impacto en la enfermedad.
- 1.
Atelectasia: su causa puede ser la presencia de un tapón mucoso intrabronquial o bien secundaria a enfermedad parenquimatosa.
- 2.
Neumotórax: se produce por rotura de bullas en la pleura visceral y aparece en un 5-8% de los pacientes, en general con enfermedad avanzada. Los síntomas de dolor torácico (precordial o irradiado al hombro) y disnea aguda nos deben hacer sospechar esta complicación.
- 3.
Hemoptisis: es muy frecuente en la evolución la hemoptisis leve o expectoración de estrías de sangre en esputo; pero en ocasiones se presentan hemoptisis moderadas o graves. Se debe a la presencia de arterias bronquiales dilatadas y tortuosas que sangran con facilidad, especialmente en el curso de las infecciones respiratorias.
- 4.
Aspergilosis broncopulmonar alérgica (ABPA): se produce como resultado de una reacción de hipersensibilidad a A. fumigatus, hongo que con frecuencia coloniza el árbol bronquial. Afecta al 1-15% de los pacientes. su diagnóstico se dificulta por la similitud de los síntomas y signos con la FQ, y se manifiesta por un deterioro clínico y funcional que no responde al tratamiento habitual. Hay unos criterios diagnósticos basados en el deterioro clínico, respuesta inmunitaria al hongo y cambios radiológicos y/o funcionales.
Lectura rápida
La edad de comienzo de los síntomas respiratorios es muy variable. A diferencia de los síntomas digestivos y la deshidratación, que pueden aparecer de forma más temprana, la clínica respiratoria puede aparecer en la primera infancia o en formas más leves, en la adolescencia o en adultos jóvenes. En el lactante, puede iniciar con clínica de tos persistente, clínica de bronquiolitis, a veces fuera de una época epidémica, o tos pertusoide.
Progresivamente, según evoluciona la enfermedad, se desarrollan bronquiectasias y aparece tos productiva. Algunos pacientes cursan con hiperreactividad bronquial y son diagnosticados de asma.
Curiosamente, los pulmones del recién nacido son histológicamente normales. Sin embargo, ya en los primeros meses de la vida algunos pacientes desarrollan una colonización bacteriana crónica endobronquial. En la fase inicial es característica la presencia de Haemophilus influenzae y/o Staphylococcus aureus. Posteriormente, la casi totalidad de los pacientes presenta colonización por Pseudomonas aeruginosa, que se asocia a un deterioro progresivo de la función pulmonar. La afectación pulmonar va a ser la que va a condicionar la mayor morbimortalidad, ya que será la causa del 95% de los fallecimientos en estos pacientes.
El diagnóstico se basa en los criterios elaborados en 1998 en el consenso promovido por la Fundación Americana para la Fibrosis Quística7, que siguen vigentes en la actualidad y que exigen, para el diagnóstico de la FQ, la existencia de, al menos, una característica fenotípica, o que un/a hermano/a hubiera sido diagnosticado/a previamente de FQ, o que el paciente tuviese un cribado neonatal positivo para FQ, más una anomalía de la conductancia transmembrana (CFTR), demostrada por: positividad del test del sudor; o identificación de una mutación causante de FQ en ambas copias de los genes que codifican la proteína CFTR, o demostración de anomalías características en el transporte iónico a través del epitelio nasal (tablas 1 y 2).
Criterios diagnósticos de fibrosis quística7
Existencia de:
|
| Más Una anomalía de la conductancia transmembrana:
|
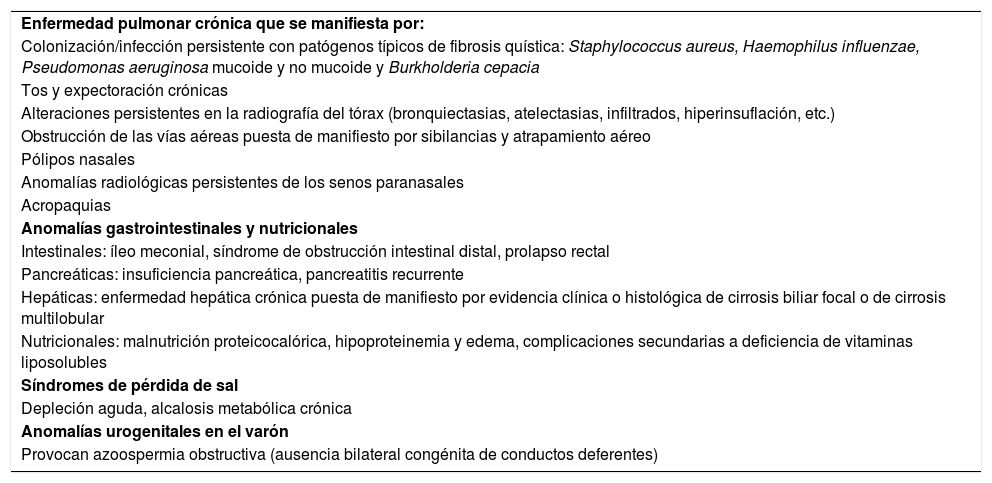
Características fenotípicas de fibrosis quística
| Enfermedad pulmonar crónica que se manifiesta por: |
| Colonización/infección persistente con patógenos típicos de fibrosis quística: Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa mucoide y no mucoide y Burkholderia cepacia |
| Tos y expectoración crónicas |
| Alteraciones persistentes en la radiografía del tórax (bronquiectasias, atelectasias, infiltrados, hiperinsuflación, etc.) |
| Obstrucción de las vías aéreas puesta de manifiesto por sibilancias y atrapamiento aéreo |
| Pólipos nasales |
| Anomalías radiológicas persistentes de los senos paranasales |
| Acropaquias |
| Anomalías gastrointestinales y nutricionales |
| Intestinales: íleo meconial, síndrome de obstrucción intestinal distal, prolapso rectal |
| Pancreáticas: insuficiencia pancreática, pancreatitis recurrente |
| Hepáticas: enfermedad hepática crónica puesta de manifiesto por evidencia clínica o histológica de cirrosis biliar focal o de cirrosis multilobular |
| Nutricionales: malnutrición proteicocalórica, hipoproteinemia y edema, complicaciones secundarias a deficiencia de vitaminas liposolubles |
| Síndromes de pérdida de sal |
| Depleción aguda, alcalosis metabólica crónica |
| Anomalías urogenitales en el varón |
| Provocan azoospermia obstructiva (ausencia bilateral congénita de conductos deferentes) |
Recientemente, se han publicado normas para que la realización del test del sudor cumpla los requisitos técnicos necesarios en todos los centros que lo realicen8 y el algoritmo diagnóstico a seguir9.
El test del sudor se considera normal cuando el cloruro es inferior a 40mEq/l, dudoso entre 40 y 60mEq/l y patológico por encima de 60mEq/l.
En la actualidad, hay programas de detección neonatal de implantación progresiva en diversos países. En España, se han iniciado desde hace unos años en algunas comunidades autónomas. Se basan en la detección de cifras elevadas de tripsina inmunorreactiva en sangre del talón del recién nacido, que se confirma con una segunda determinación a los 28 días de vida o un estudio genético. En el caso de que este cribado sea positivo, se confirmará con el test de sudor.
También es posible el diagnóstico prenatal por biopsia corial o amniocentesis.
Lectura rápida
El diagnóstico se basa en los criterios elaborados en 1998 en el consenso promovido por la Fundación Americana para la Fibrosis Quística, que siguen vigentes en la actualidad y que exigen para el diagnóstico de FQ la existencia de, al menos, una característica fenotípica, o que un/a hermano/a haya sido diagnosticado/a previamente de FQ, o que el paciente tenga un cribado neonatal positivo para FQ más una anomalía de la conductancia transmembrana (CFTR) demostrada por: positividad del test del sudor; identificación de una mutación causante de FQ en ambas copias de los genes que codifican la proteína CFTR, o demostración de anomalías características en el transporte iónico a través del epitelio nasal.
La FQ precisa de un tratamiento integral y multidisciplinario, por ser una enfermedad muy compleja y afectar a varios órganos. En la actualidad se realiza en unidades multidisciplinarias organizadas en grupos de trabajo, donde además se contempla el apoyo psicológico. Esto ha supuesto uno de los principales factores favorecedores de la buena evolución de estos pacientes y el aumento de sus expectativas de vida10.
El objetivo del tratamiento de la afectación pulmonar11,12 deberá enfocarse en la prevención y el enlentecimiento del deterioro pulmonar, con la mejora de los síntomas de obstrucción, inflamación e infección13.
Medidas generalesComo medidas generales se aconseja a los pacientes recibir el calendario vacunal y la vacuna antigripal todos los otoños, evitar el tabaquismo activo y pasivo, evitar la exposición a las infecciones virales (guardería, pacientes, etc.) e infecciones respiratorias, evitar la exposición a hongos ambientales (establos, abonos, etc.) y lograr un estado nutricional óptimo (adecuada nutrición, suplementos vitamínicos y enzimas).
Tratamiento de la obstrucciónPara prevenir y mejorar la obstrucción, se recomienda realizar fisioterapia respiratoria diaria y deporte. La fisioterapia se debe adaptar a la edad del paciente y se aconseja realizarla 2 veces al día, intensificándose en las reagudizaciones. El deporte se deberá adaptar a la afectación cardiopulmonar en cada momento, y su objetivo es actuar como fisioterapia, con el incremento de la capacidad pulmonar y la fuerza muscular con un importante papel de relación social.
Los broncodilatadores inhalados de acción corta se administran antes de realizar la fisioterapia respiratoria, si se ha demostrado mejoría espirométrica tras su aplicación y en los procesos de hiperreactividad bronquial.
Se ha demostrado que los mucolíticos desoxirribonucleasa (DNasa) recombinante (Pulmozyme®) destruyen el ADN liberado en la inflamación (éste aumenta la viscosidad de las secreciones), lo que facilita la eliminación de las secreciones14. Se aplica en aerosol tras los broncodilatadores y la fisioterapia.
El suero salino hipertónico en aerosol parece tener una eficacia similar15,16.
Tratamiento de la inflamaciónLos corticoides orales sólo se utilizan en ocasiones muy concretas: en hiperreactividad bronquial importante, ABPA y, a veces, como complemento para disminuir la inflamación crónica de las vías aéreas17.
El ibuprofeno se ha empleado en algunos estudios con resultados variables18.
Los antileucotrienos precisan de estudios que muestren su beneficio terapéutico.
Actualmente, los macrólidos se utilizan por sus propiedades inmunomoduladoras, y por interferir en la formación del biofilm producido por P. aeruginosa. En la actualidad, se está generalizando su uso en pacientes mayores de 6 años crónicamente colonizados por P. aeruginosa. Se administra azitromicina 500mg 3 veces a la semana en pacientes de más de 40kg y dosis de 250mg si es un paciente de menos de 40kg19.
Tratamiento de la infección respiratoriaEl tratamiento antibiótico, junto con la fisioterapia respiratoria, son la base fundamental del tratamiento en la FQ La elección del antibiótico adecuado se realiza mediante el estudio microbiológico periódico de las secreciones respiratorias (esputo, aspirado nasal o frotis faríngeo). Según los resultados, se indica el tratamiento antimicrobiano en las exacerbaciones20.
Tratamiento temprano en el primer aislamiento de Pseudomonas aeruginosaSe recomienda el tratamiento temprano e intenso para erradicar o retrasar la colonización crónica. se realizará tratamiento oral con ciprofloxacino durante 3–4 semanas, pero si el paciente no está estable, se llevará a cabo tratamiento intravenoso con betalactámico y aminoglucósido durante 2–3 semanas. Al mes del inicio del tratamiento, se debe repetir el cultivo, y si es positivo se debe repetir otro ciclo de antibiótico sistémico. Y si tras este 2.° ciclo sigue siendo positivo, se actuará como en la colonización crónica. se debe asociar también tratamiento inhalado con colimicina o tobramicina de forma continua, que se mantendrá 6–12 meses21.
Tratamiento de las exacerbacionesAnte cualquier signo de empeoramiento de la enfermedad respiratoria (aumento de la tos, expectoración, cambio en la consistencia o color de éstas, disminución de la función pulmonar, etc.) y aunque no tenga fiebre, se debe administrar un ciclo de antibióticos de entre 2 y 3 semanas de duración, por vía oral o intravenosa, según la afectación del paciente y el patógeno de colonización habitual (según los cultivos periódicos realizados). si se emplea la vía intravenosa, se recomienda asociar 2 antimicrobianos para limitar las resistencias. La modalidad de tratamiento intravenoso domiciliario es posible si se cumplen unos criterios médicos de efectividad y las condiciones personales y familiares lo permiten.
Tratamiento crónico de mantenimientoEl objetivo es reducir la carga bacteriana y enlentecer el círculo infección-inflamación.
El tratamiento inhalado de antibióticos de forma crónica ha demostrado una mejoría en la función pulmonar, en el número de reagudizaciones y de ingresos, lo que mejora la calidad de vida. Los fármacos más empleados son la colimicina (prácticamente no hay cepas de Pseudomonas resistentes a ella) y tobramicina, que se administran nebulizados22.
Tratamiento de las complicaciones respiratorias no infecciosasAtelectasiaSi la causa es un tapón de moco intrabronquial, se tratará con antibióticos intravenosos, broncodilatadores, mucolíticos y fisioterapia respiratoria intensiva. si no hay respuesta al tratamiento, se puede realizar una fibrobroncoscopia para aspirar las secreciones espesas e instilar DNasa localmente.
NeumotóraxEl paciente debe ser siempre enviado al hospital ante la sospecha. En caso de neumotórax pequeño, se trata con medidas conservadoras (ingreso, reposo y oxigenoterapia). Si es sintomático o de tamaño superior, se tratará con tubo torácico de drenaje y oxigenoterapia y si no se resuelve o es de repetición, se recomienda pleurodesis quirúrgica.
HemoptisisLa hemoptisis leve suele ser signo de exacerbación respiratoria, o bien por el uso de fármacos antiinflamatorios no esteroideos o déficit de vitamina K. El tratamiento será corregir la causa precipitante, administrar antibióticos en caso de exacerbación pulmonar, evitar la fisioterapia respiratoria intensiva y retirar la medicación inhalada.
En caso de hemoptisis importante, se realizará ingreso hospitalario, y se mantendrá la permeabilidad respiratoria y la estabilidad hemodinámica. Algunos pacientes precisan embolización de las arterias bronquiales que debe realizarse en centros con experiencia.
Aspergilosis broncopulmonar alérgicaEl tratamiento de elección son los corticoides orales: prednisolona a 0,5-2mg/kg/día (máximo 60mg/día), con descenso progresivo durante 6 meses, y valoración de la respuesta clínica y analítica. A veces se asocia itraconazol.
Lectura rápida
El objetivo del tratamiento de la afectación pulmonar deberá enfocarse en la prevención y el enlentecimiento del deterioro pulmonar, y en mejorar los síntomas de obstrucción, con broncodilatadores, fisioterapia y agentes mucolíticos (desoxirribonucleasa y suero salino hipertónico), la inflamación (ibuprofeno, corticoides y azitromicina que se emplea por su efecto inmunomodulador de forma continua en colonizaciones crónicas por P. aeruginosa), y la infección con antibióticos sistémicos y, en algunos casos, nebulizados (colimicina o tobramicina). Se debe insistir en un tratamiento enérgico de la primera colonización por P. aeruginosa. En la actualidad hay nuevos tratamientos en fase de investigación.
El trasplante pulmonar sería, por último, el único procedimiento terapéutico en la enfermedad pulmonar avanzada.
Algunos pacientes deberán recibir tratamiento con oxigenoterapia si presentan desaturaciones. El apoyo ventilatorio con ventilación no invasiva es de gran ayuda de forma puntual en el tratamiento de las reagudizaciones en algunos pacientes, y de forma crónica en otros pacientes en espera de trasplante pulmonar23.
Trasplante pulmonarSe considera una importante opción terapéutica cuando la función pulmonar está muy afectada y la calidad de vida, muy deteriorada24, por lo que se indica en pacientes menores de 65 años, con enfermedad pulmonar avanzada sintomática, con una esperanza de vida menor de 2 años y con ausencia de contraindicaciones.
La supervivencia es del 73% en el primer año, del 57% a los 3 años y del 45% a los 5 años25.
Tratamiento génicoEl objetivo es transferir una copia normal del gen a las células de los pacientes con FQ,para restablecer la función celular normal. Su aplicación sería curativa, pero es muy complejo y aún no se ha podido conseguir.
Restauración del transporte iónicoEn la actualidad están en fase de investigación diversos tratamientos que intentan restaurar el transporte de iones a través de vías no dependientes de CFTR y que representan una alternativa atractiva de tratamiento en la FQ: denufosol tetrasódico MOLI1901, SPI-8811 y el INO4995.
En conclusión, gracias a los nuevos tratamientos y al tratamiento más radical de la enfermedad pulmonar, se ha obtenido una gran mejoría en la calidad de vida y en la supervivencia de estos pacientes, y se mantiene la esperanza de que en un futuro no muy lejano se pueda encontrar un tratamiento curativo de esta enfermedad.