














Puntos clave

La dermatomiositis juvenil es una enfermedad rara, de naturaleza autoinmunitaria e inicio antes de los 16 años. Se trata de una vasculopatía caracterizada por debilidad muscular proximal simétrica, elevación sérica de las enzimas musculares y lesiones cutáneas patognomónicas que incluyen el eritema heliotropo y las pápulas de Gottron. Se clasifica dentro de las miopatías inflamatorias idiopáticas; afecta principalmente a los músculos y la piel mediante la inflamación de pequeños vasos, pero puede afectar a cualquier órgano. Clásicamente, el pronóstico de la dermatomiositis juvenil era malo, con mortalidad en una tercera parte de los pacientes y otro tercio con afectación funcional permanente1. En las últimas décadas, con la introducción de nuevos tratamientos y el diagnóstico más precoz de esta entidad la mortalidad ha disminuido de forma drástica y ha mejorado el pronóstico funcional de estos pacientes2.
EpidemiologíaLa incidencia anual de la enfermedad en EE.UU. es de 3,2 por millón de niños menores de 17 años3. La edad media al inicio es de 7 años, siendo el 25% de los pacientes menores de 4 años al manifestarse la enfermedad4. Predomina en el sexo femenino; la proporción mujer:varón en EE.UU. es de 2,3:14 y en Inglaterra de 5:15. En la actualidad no se dispone de registros sobre incidencia y prevalencia en la población pediátrica española.
EtiopatogeniaLa etiopatogenia de la dermatomiositis juvenil es desconocida. Se han sugerido diferentes mecanismos patogénicos, incluidos factores genéticos e inmunológicos predisponentes y el papel de algunos desencadenantes como las infecciones y la luz ultravioleta. Al igual que en otras enfermedades autoinmunes, probablemente resulta de la interacción entre factores ambientales en un individuo genéticamente predispuesto.
A diferencia de los pacientes adultos, los niños afectados no tienen un mayor riesgo de neoplasia6.
Factores genéticosEntre los factores genéticos se ha descrito la asociación a determinados alelos HLA (B8, DRB1*0301, DQA1*0501 y DQA1*0301) y a algunos polimorfismos en el gen del factor de necrosis tumoral alfa (TNF-α), que condicionarían una mayor producción de esta citocina proinflamatoria7.
Factores ambientalesLa presencia de un desencadenante infeccioso se sospecha por 3 observaciones:
- —
La aparente estacionalidad en el inicio de la enfermedad (con predominio en verano y primavera).
- —
La distribución estacional en las fechas de nacimiento de los pacientes, lo cual sugiere una influencia de las exposiciones en etapas precoces de la vida para el desarrollo posterior de la enfermedad8.
- —
La descripción de que muchos pacientes afectados tienen el antecedente de infecciones del tracto respiratorio superior o gastrointestinal en los meses previos9. No obstante, hasta la fecha no se ha identificado ningún microorganismo responsable7. La presencia de fotosensibilidad en esta enfermedad sugiere que la luz ultravioleta podría tener también un papel en el desarrollo de la enfermedad10.
Lectura rápida

La dermatomiositis juvenil es una enfermedad rara, de naturaleza autoinmunitaria e inicio en menores de 16 años. Se caracteriza por debilidad muscular proximal simétrica, elevación sérica de las enzimas musculares y lesiones cutáneas patognomónicas.
EtiopatogeniaEs desconocida, pero probablemente resulta de la interacción entre factores ambientales (infecciones, luz ultravioleta) en un individuo genéticamente predispuesto. A diferencia de los pacientes adultos, los niños con dermatomiositis juvenil no tienen un mayor riesgo de neoplasia.

Se ha postulado la influencia de factores perinatales en el desarrollo de la enfermedad: la transferencia de células maternas hacia el feto durante el embarazo, llamado microquimerismo materno, se puede observar hasta en un 80-100% de muestras de tejido muscular de pacientes con dermatomiositis juvenil11. Estas células maternas podrían desarrollar una inmunorreactividad hacia los tejidos propios del individuo, y conducir a la enfermedad.
La combinación de factores predisponentes y desencadenantes llevaría a una activación del sistema inmunitario que por diferentes mecanismos (infiltración de linfocitos B, T y células dendríticas en el músculo con lesión capilar mediada por complemento) produce una vasculitis inflamatoria. Esta vasculitis sería la responsable de las manifestaciones clínicas de la enfermedad. La inflamación vascular no está limitada a los vasos del músculo y la piel, sino que también puede afectar a otros tejidos como tracto gastrointestinal, pulmones, ojos, riñones y corazón, y condicionar las diferentes manifestaciones y complicaciones de la enfermedad.
Manifestaciones clínicasSignos y síntomas constitucionalesAl manifestarse la enfermedad, los pacientes frecuentemente presentan malestar general, fatiga, anorexia e irritabilidad, que por lo general se atribuye a una virasis. En algunas series se describe la presencia de fiebre hasta en un 20% de los pacientes al inicio de la enfermedad12. Los cambios en el patrón de la marcha en este estadio precoz de la enfermedad deben hacer sospechar la afectación muscular. Posteriormente aparecerán las manifestaciones cardinales de la enfermedad como los exantemas, debilidad manifiesta y, en algunos pacientes, calcinosis.
Manifestaciones musculoesqueléticasLa dermatomiositis juvenil se caracteriza por la debilidad muscular. El inicio suele ser insidioso con empeoramiento progresivo a lo largo de varios meses; sólo en algunos casos se describe un inicio agudo. Aunque esta debilidad puede afectar a cualquier grupo muscular es más manifiesta a nivel proximal (cintura escapular y pélvica) y axial, en especial en la musculatura de hombros, caderas, flexores de la cabeza y musculatura abdominal, y habitualmente es simétrica y parcheada. Los músculos afectados pueden estar edematosos, indurados o dolorosos. En la exploración pueden apreciarse los signos de Gowers y Trendelemburg. La afectación de la musculatura distal tiende a ser más leve y habitualmente no conlleva una alteración funcional significativa. Hasta una cuarta parte de los pacientes pueden tener afectación de la musculatura faríngea, hipofaríngea y palatina12, que se manifiesta en forma de disfonía, dificultades en iniciar la deglución, disfagia y voz nasal. En estos casos hay un riesgo elevado de broncoaspiración.
Se ha descrito la presencia de artralgias y artritis leve, no deformante ni erosiva13. Aparece habitualmente al inicio de la enfermedad y responde bien al tratamiento habitual de la dermatomiositis juvenil. No obstante, en presencia de una artritis significativa debería considerarse la presencia de un síndrome de superposición (con características de lupus, artritis idiopática juvenil o esclerodermia).
La evaluación de la fuerza muscular en estos pacientes debe registrarse mediante el uso de escalas estandarizadas que evalúan grupos musculares seleccionados por separado; una de las más utilizadas es la Escala de valoración de miositis en niños o CMAS (Childhood Myositis Assessment Scale)14.
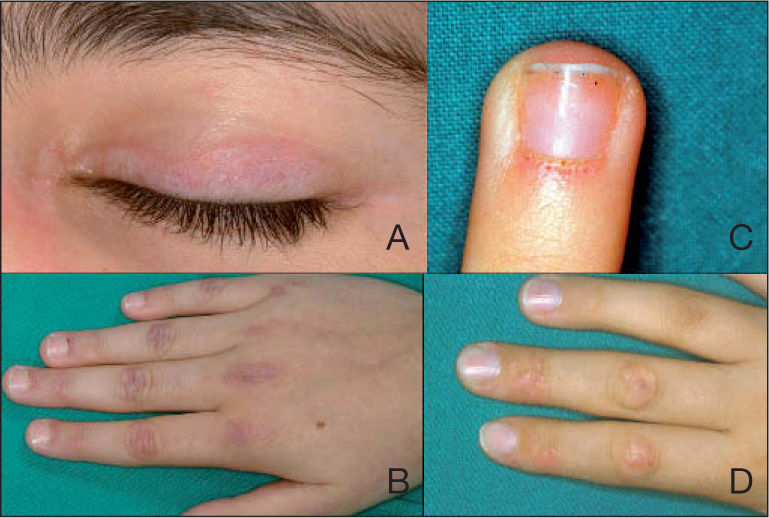
Manifestaciones cutáneasLas manifestaciones cutáneas son variadas y constituyen la clave para el diagnóstico de la enfermedad. Pueden ser autolimitadas y aparecer antes o después del inicio de la debilidad. Las manifestaciones más características (eritema heliotropo y pápulas de Gottron) son patognomónicas y se observan hasta en el 80% de los pacientes.
- —
Eritema heliotropo (fig. 1A): consiste en un exantema eritemato-violáceo localizado en los párpados superiores, generalmente acompañado de edema palpebral. Es el signo más específico de la enfermedad pero no siempre está presente. Algunos pacientes también desarrollan telangiectasias en párpados y eritema malar que, a diferencia del observado en el lupus, no parece respetar los pliegues nasolabiales.
- —
Pápulas de Gottron (figs. 1B y D): son la manifestación más común. Se trata de pápulas rosadas o violáceas localizadas en la superficie extensora de los nudillos (metacarpofalángicas, interfalángicas proximales y distales), codos, rodillas y maléolo medial. Generalmente respeta los espacios interfalángicos. El signo de Gottron consiste en un exantema en las mismas localizaciones pero no palpable (macular). A veces las lesiones pueden ser escamosas y secas, simulando una psoriasis. Con el tiempo pueden desarrollar un centro atrófico y blanquecino con telangiectasias.
- —
Las alteraciones en los capilares periungueales (fig. 1C) son características y frecuentes en la dermatomiositis juvenil15 y reflejan la presencia de una vasculopatía sistémica. Puede observarse eritema periungueal, hipertrofia de la cutícula, pérdida y dilatación de los capilares periungueales y pequeñas erosiones superficiales12.
- —
Otras presentaciones menos frecuentes consisten en un eritema en forma de V en la parte superior del tronco o difuso en tronco y extremidades, que puede ser descamativo y evolucionar hacia la formación de telangiectasias. Generalmente la exposición solar agrava estos exantemas. Debido a la presencia de una vasculitis sistémica también pueden aparecer ulceraciones en axilas, hombros, canto interno del ojo o en zonas de presión. Los pacientes con exantema generalizado y ulceraciones cutáneas al inicio tienen un peor pronóstico, ya que esta ulceración refleja una vasculopatía más extensa16.

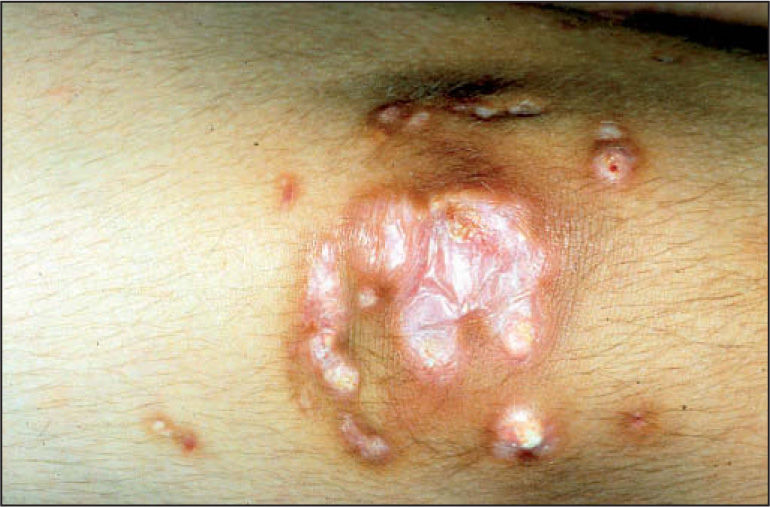
La calcificación de tejidos blandos (fig. 2) puede producirse entre el 20 y el 40% de los casos de dermatomiositis juvenil y se relaciona con la gravedad de la enfermedad cutánea, la presencia de vasculopatía y el retraso en el inicio del tratamiento17.

Aparecen en cualquier localización pero generalmente se encuentran en codos, rodillas, partes acras y zonas de traumatismos. Pueden ocasionar dolor local, contracturas articulares, ulceración de la piel adyacente y condicionan un peor pronóstico funcional. No suelen observarse en el momento del diagnóstico sino que aparecen más tardíamente en el curso de la enfermedad. Pueden apreciarse pequeños nódulos o placas superficiales, depósitos profundos seudotumorales, depósitos difusos en los planos miofasciales y, en casos excepcionales, calcificaciones generalizadas que conforman un exosqueleto.
Afectación gastrointestinalConstituye una de las complicaciones más graves de la enfermedad y puede afectar a la supervivencia. Es debida a la presencia de vasculitis gastrointestinal que conduce a ulceraciones mucosas por oclusiones vasculares. Clínicamente se presenta como dolor abdominal, pancreatitis, sangrado gastrointestinal o perforación18.
LipodistrofiaRaramente observada al diagnóstico, aparece en el curso de la enfermedad en el 14-25% de los pacientes19,20. La lipodistrofia se caracteriza por una pérdida lenta y progresiva del tejido subcutáneo que afecta de forma mayoritaria al segmento superior del cuerpo. Por lo general se asocia a acantosis nigricans, hipertrigliceridemia y resistencia a la insulina. Su origen probablemente es multifactorial, y se incluyen los cambios metabólicos ocasionados por la inflamación muscular y el tratamiento prolongado con corticoides.
Lectura rápida
Las manifestaciones cutáneas son claves para el diagnóstico. El eritema heliotropo y las pápulas de Gottron son patognomónicos de dermatomiositis juvenil. La debilidad muscular es simétrica y de predominio proximal, afectando a la cintura escapular y pélvica. Debe vigilarse la aparición de disfagia y debilidad en la musculatura respiratoria. La afectación gastrointestinal constituye una de las complicaciones más graves con compromiso vital.
Las calcificaciones distróficas pueden aparecer en un 40% de los pacientes y se relacionan con la presencia de enfermedad grave y un tratamiento insuficiente o de inicio tardío.
La afectación pulmonar es mucho menos frecuente en niños que en adultos y puede manifestarse como una enfermedad intersticial pulmonar. Con más frecuencia se observa debilidad respiratoria, que es responsable de la presencia de síntomas respiratorios en hasta un tercio de los pacientes19. También debe recordarse que la disfagia puede ocasionar broncoaspiraciones con el consiguiente daño pulmonar, y que los tratamientos inmunosupresores recibidos pueden facilitar las infecciones pulmonares oportunistas.
En el corazón pueden detectarse soplos, cardiomegalia o pericarditis. La presencia de afectación cardíaca grave (miocarditis, alteraciones en la conducción o bloqueos) es rara, pero puede aparecer años después del inicio de la enfermedad18,21.
Otras manifestacionesEl inicio de la enfermedad en forma de anasarca es un signo de mal pronóstico, y se debe a una pérdida capilar difusa por lesión endotelial en los vasos22. La presencia de vasculitis en otros órganos (vejiga urinaria, vagina, testes o sistema nervioso), a pesar de ser rara, también está asociada a un peor pronóstico.
Dermatomiositis amiopáticaEs una entidad rara que consiste en la presencia de los exantemas característicos de la dermatomiositis juvenil pero sin afectación muscular durante más de 6 meses. Con más frecuencia se da el caso de que los pacientes presenten una afectación muscular leve que pasa desapercibida. En general estos pacientes tienen un buen pronóstico23,24, a pesar de que también pueden desarrollar calcinosis y artritis.
Lectura rápida
Para el diagnóstico se utilizan los criterios de Bohan y Peter.
El electromiograma muestra un patrón miopático con denervación y es de utilidad para determinar el origen muscular de la debilidad. En las pruebas de laboratorio generalmente se observa elevación de una o varias de las enzimas musculares (creatincinasa, lactato deshidrogenasa, aldolasa, aspartato aminotransferasa).
La biopsia muscular es la prueba definitiva para establecer el diagnóstico de dermatomiositis juvenil, demostrando la presencia de una vasculitis en el tejido muscular. El uso de nuevas tecnologías como la resonancia magnética podría replantear la necesidad de la biopsia para el diagnóstico de dermatomiositis juvenil.
El diagnóstico de la dermatomiositis juvenil se realiza a través de los criterios, descritos en 1975, de Bohan y Peter25,26 (tabla 1). Según el número de criterios que presente el paciente, se diagnosticará como definida, probable o posible. Su sensibilidad y especificidad (a pesar de que no han sido validadas en niños) serían, respectivamente, de alrededor del 45–90 y del 90%18.
Criterios de Bohan y Peter para el diagnóstico de dermatomiositis juvenil
|
| Diagnóstico de dermatomiositis juvenil: presencia del primer criterio y al menos 2 de los otros 4 criterios. Se diferencia entre: |
| Definida: 3 de los otros 4 criterios |
| Probable: 2 de los otros 4 criterios |
| Posible: 1 de los otros 4 criterios |
AST: aspartato aminotransferasa; CK: creatincinasa; EMG: electromiograma; LDH: lactato deshidrogenasa.
Los cambios en la práctica clínica a lo largo de los años han incorporado el uso de nuevas técnicas diagnósticas no invasivas, como la resonancia magnética (RM) en lugar del electromiograma y la biopsia muscular. Por este motivo, un grupo colaborativo internacional está realizando una revisión de los criterios diagnósticos para adaptarlos a la práctica clínica actual27.
Laboratorio- —
Enzimas musculares: las enzimas musculares utilizadas habitualmente para la evaluación de la lesión muscular son la creatincinasa (CK), lactato deshidrogenada (LDH), aldolasa y aspartato aminotransferasa (AST).
Generalmente se elevan uno o más de estos parámetros, pero en fases precoces de la enfermedad pueden mantenerse en valores normales hasta que se desarrolla la debilidad clínicamente aparente.
- —
Autoanticuerpos: en el 60% de los pacientes pueden detectarse anticuerpos antinucleares positivos. Éstos no son específicos de la enfermedad, por lo que no tienen valor diagnóstico. En el 30% de adultos con miositis se han descrito una serie de autoanticuerpos específicos que estarían asociados con determinados subgrupos clínicos de la enfermedad. En niños también se ha descrito la asociación con algunos autoanticuerpos aunque en menor frecuencia. Entre estos destacan los anti-Mi-2 (5% de los casos y asociado a buena respuesta al tratamiento), antisintetasa (5-10%, asociado a un curso agudo y con posibles complicaciones como artritis o enfermedad intersticial pulmonar), y recientemente se han descrito los anti-p155 (20-30% de pacientes). Actualmente en los laboratorios clínicos aún no está disponible la determinación de estos anticuerpos, por lo que se sólo se realiza con finalidad de investigación10.
- —
Otros parámetros: el hemograma y la velocidad de sedimentación globular habitualmente son normales, por lo que no son de demasiada utilidad para el control de la enfermedad28. En pacientes con enfermedad activa pueden encontrarse elevaciones del factor Von Willebrand, que reflejaría la lesión endotelial29.
El electromiograma (EMG) muestra un patrón miopático (potenciales motores polifásicos de baja amplitud y corta duración) con denervación (fibrilaciones espontáneas, ondas positivas y descargas de alta frecuencia)18. Estos cambios no son específicos de dermatomiositis juvenil, por lo que el EMG sólo es útil para confirmar que el origen de la debilidad es muscular. En algunos pacientes con dermatomiositis juvenil activa el EMG puede ser normal (probablemente por la naturaleza parcheada de la inflamación muscular); por este motivo es recomendable realizar el registro electromiográfico en diferentes localizaciones.
Biopsia muscularEs la prueba definitiva para establecer el diagnóstico de dermatomiositis juvenil. Es preferible realizar una biopsia abierta que una biopsia por punción, para así preservar la orientación de las fibras y obtener una muestra de más calidad. El rasgo característico de la dermatomiositis juvenil es la presencia de una vasculopatía, fundamentalmente piel, músculo y tracto gastrointestinal. Los cambios histológicos en el músculo incluyen edema del endotelio capilar con obliteración del lumen, atrofia e inflamación perifascicular y fenómenos de degeneración y regeneración muscular.
Lectura rápida
Consiste en la administración de corticoides e inmunosupresores durante un período largo de tiempo (1–3 años), con el objetivo de alcanzar y mantener la remisión y prevenir las complicaciones. El metotrexato es el inmunosupresor más utilizado como ahorrador de corticoides. El tratamiento con fisioterapia y terapia ocupacional es de gran importancia para la rehabilitación y prevención de complicaciones.
El mejor tratamiento para las calcinosis es la prevención de su formación mediante un tratamiento precoz y agresivo de la dermatomiositis juvenil. Una vez instaurada la calcinosis, ninguna terapia ha demostrado ser de utilidad.
Los fármacos biológicos (antifactor de necrosis tumoral [TNF], rituximab) se han ensayado con resultados variables. La ciclofosfamida se utiliza en casos con grave compromiso vital.

Estas nuevas tecnologías tienen un interés creciente en el diagnóstico y seguimiento de esta enfermedad. La RM muscular puede demostrar áreas de inflamación muscular y, a diferencia del EMG, permite estudiar amplias áreas musculares. Además, al tratarse de una prueba no invasiva, tiene la ventaja de poder realizar estudios sucesivos para seguir el curso de la enfermedad. También puede utilizarse para seleccionar las zonas afectadas donde realizar la biopsia muscular, aumentando así la rentabilidad diagnóstica de la biopsia.
Los hallazgos observados en pacientes con dermatomiositis juvenil activa en secuencias T2 con supresión grasa y en secuencias STIR (short T1 inversion recovery) son un aumento de señal en los músculos afectados, edema perimuscular y una hiperintensidad en el tejido graso subcutáneo. Se visualiza como zonas blancas dentro de los músculos donde sólo debería haber imágenes oscuras, reflejando el edema debido a la inflamación30. Tras la remisión de la enfermedad estos cambios desaparecen.
La RM con espectroscopia proporciona un estudio del metabolismo muscular. Es una prueba de gran sensibilidad que detecta el descenso en el metabolismo de los músculos afectados31. En el futuro parece probable que estas técnicas se implementen en los procesos de diagnóstico y manejo habituales de los pacientes con dermatomiositis juvenil, pudiendo incluso reemplazar en algunos casos al EMG y la biopsia.
Diagnóstico diferencialEn fases iniciales el diagnóstico es difícil por la naturaleza poco específica de sus síntomas, que pueden confundirse con un proceso viral. Una vez instaurada la enfermedad, en función de las características clínicas predominantes el diagnóstico diferencial deberá realizarse con gran variedad de procesos7 (tabla 2).
Diagnóstico diferencial en la dermatomiositis juvenil
| Signo guía | Diagnóstico diferencial |
|---|---|
| Debilidad | Distrofias musculares |
| Miopatías metabólicas (glucogenosis, lipidosis, enfermedades mitocondriales) | |
| Miopatías endocrinas (hiper o hipotiroidismo, enfermedad de Cushing, diabetes mellitus) | |
| Miopatía inducida por fármacos (corticoides, interferón alfa, estatinas, hidroxiclorocina, amfotericina B, cimetidina, vincristina) | |
| Enfermedades neuromusculares (miastenia gravis) | |
| Enfermedades de la motoneurona (atrofia muscular espinal) | |
| Debilidad con o sin exantemas | Infecciones virales (Enterovirus, Influenza, Coxsackie, Echovirus, Parvovirus, Poliovirus, hepatitis B) |
| Infecciones bacterianas y parasitarias (estafilococos, estreptococos, toxoplasmosis, triquinosis, enfermedad de Lyme) | |
| Enfermedades inflamatorias (celiaquía, enfermedad inflamatoria intestinal) | |
| Otras enfermedades reumáticas (lupus eritematoso sistémico, esclerodermia, artritis idiopática juvenil, enfermedad mixta del tejido conjuntivo, vasculitis idiopáticas) | |
| Exantema sin debilidad | Eccemas, psoriasis y alergias |
Dada la baja frecuencia de esta enfermedad, el tratamiento de la dermatomiositis juvenil no ha sido avalado por estudios controlados aleatorizados sino que se basa en el conocimiento derivado de la experiencia clínica y estudios observacionales. Generalmente consiste en la administración de corticoides e inmunosupresores durante un período largo de tiempo (1–3 años), con el objetivo de alcanzar y mantener la remisión y prevenir las complicaciones. La respuesta al tratamiento se determina mediante la evaluación seriada de signos y síntomas clínicos (presencia de exantemas, dolor muscular, fatiga), datos de laboratorio (enzimas musculares), fuerza muscular (mediante escalas de valoración como el CMAS) y, opcionalmente, otros estudios (RM muscular)18.
CorticoidesDesde 1970 el tratamiento estándar de la dermatomiositis juvenil han sido los corticoides a dosis de 1–2 mg/kg/día por vía oral mantenidos hasta alcanzar una mejoría clínico-analítica, y posteriormente en dosis descendientes durante un período de 2 años. No obstante, para evitar los efectos secundarios que conllevan estos fármacos a dosis altas y de forma prolongada, en la actualidad muchos pacientes son tratados de forma precoz con inmunosupresores y/o dosis altas de metilprednisolona (30 mg/kg/día, con un máximo de 1 g/día)32, lo cual permite una reducción más rápida de los corticoides, con buena respuesta terapéutica33,34.
InmunosupresoresSon utilizados como agentes ahorradores de corticoides, y de entre ellos el metotrexato es el más utilizado. Se administra en dosis de 10–15 mg/m2 por semana, por vía oral o subcutánea, generalmente al inicio de la enfermedad. Estudios comparativos con pacientes que sólo recibieron corticoides han demostrado que la introducción precoz de metotrexato reduce las secuelas y permite alcanzar el mismo control de la enfermedad pero con la mitad de dosis acumulativa de corticoides, lo que conlleva un menor aumento de peso y una mejoría en la velocidad de crecimiento34.
Lectura rápida
El curso clínico puede ser monocíclico, policíclico o crónico persistente. Los indicadores de alto riesgo incluyen la presencia de ulceraciones en piel, tracto gastrointestinal y la enfermedad pulmonar.
PronósticoLa introducción de los corticoides en el tratamiento ha cambiado espectacularmente el pronóstico de la enfermedad: de una mortalidad del 33% y cronicidad con secuelas persistentes en otro 33%, se ha pasado a una supervivencia > 90% y un buen pronóstico funcional. Un tratamiento insuficiente o su inicio tardío es uno de los predictores más importantes de mal pronóstico.
La ciclosporina también se ha utilizado como ahorrador de corticoides, pero tiene el inconveniente de producir hipertensión e hirsutismo. Actualmente está en curso un estudio aleatorizado multicéntrico para comparar el tratamiento con corticoides solos con el tratamiento combinado corticoides-metotrexato y corticoides-ciclosporina35.
Inmunoglobulinas intravenosasSuelen emplearse como tratamiento adyuvante en casos de resistencia o dependencia de los corticoides, sobre todo en la enfermedad cutánea resistente. Se administran en dosis de 2 g/kg cada 2–4 semanas36, y habitualmente son bien toleradas.
CiclofosfamidaSe puede utilizar en el tratamiento de pacientes graves con alto riesgo de morbimortalidad. Los indicadores de alto riesgo incluyen la presencia de ulceraciones en piel y tracto gastrointestinal, y la enfermedad pulmonar37.
Agentes biológicosLos fármacos anti-TNF se utilizan en el tratamiento de diferentes enfermedades reumatológicas. Dada la evidencia del papel del TNF-α en la fisiopatología de la dermatomiositis juvenil, se ha ensayado el tratamiento con estos fármacos (en concreto infliximab y etanercept) con resultados variables38,39. El rituximab, un anticuerpo monoclonal antilinfocito B, se ha utilizado en algunos estudios con resultados favorables40.
Otros agentes de segunda líneaSe tiene experiencia en el uso de otros fármacos como azatioprina, tacrolimus sistémico41 y tópico. Específicamente para el tratamiento de las manifestaciones cutáneas se utilizan hidroxiclorocina y micofenolato mofetilo.
Terapias adyuvantesLa fisioterapia y la terapia ocupacional son de gran importancia para la rehabilitación de los pacientes, sobre todo en aquellos que presentan secuelas con afectación funcional o mayor debilidad. Históricamente se creía que el ejercicio podía causar lesión muscular potenciando la inflamación en este nivel, pero en la actualidad se reconoce su beneficio para aumentar la fuerza muscular y la capacidad aeróbica42.
Dada la presencia de fotosensibilidad en la dermatomiositis juvenil debe utilizarse fotoprotección de forma sistemática para minimizar la aparición de exantemas y enfermedad cutánea. También es de importancia el cuidado de la piel mediante cremas emolientes y apósitos protectores en caso de ulceraciones cutáneas.
En casos de debilidad grave debe buscarse la posible presencia de disfagia y afectación respiratoria. En estos casos está indicado valorar la alimentación por sonda nasogástrica y la necesidad de soporte respiratorio.
Tratamiento de las complicaciones (calcinosis y osteoporosis)El tratamiento con corticoides es prolongado, por lo que se recomienda la administración de suplementos de calcio y vitamina D para evitar la pérdida de densidad mineral ósea43.
Para la calcinosis se han ensayado múltiples terapias como el diltiazem, hidróxido de aluminio, probenecid, bifosfonatos e inyección local de corticoides, entre otros. Ninguno de ellos ha demostrado ser eficaz36, por lo que es importante la prevención de esta complicación mediante un tratamiento precoz y agresivo de la dermatomiositis juvenil. En casos graves se recomienda la exéresis quirúrgica, pero la calcinosis puede recurrir si la actividad de la enfermedad no está controlada44.
Evolución y pronósticoAntes de la introducción del tratamiento con corticoides la enfermedad tenía muy mal pronóstico: una tercera parte de los pacientes moría; otra tercera parte presentaba enfermedad progresiva con secuelas graves y persistencia de enfermedad crónica activa en la edad adulta45; y otro tercio se recuperaban. Actualmente el pronóstico de la dermatomiositis juvenil es generalmente bueno2: la supervivencia es muy alta, superior al 90%, con resultados funcionales buenos2,18.
El curso clínico puede ser monocíclico (alcanzando la remisión mantenida a los 2–3 años), policíclico (períodos de remisión seguidos de recidivas), y crónico persistente2,46. Un tratamiento insuficiente o su inicio tardío es uno de los predictores más importantes de mal pronóstico, con enfermedad crónica persistente y complicaciones como el retraso en el crecimiento, calcificaciones y enfermedad cutánea más extensa17,47. El diagnóstico y tratamiento precoces de la dermatomiositis juvenil, por lo tanto, constituyen la clave para mejorar el pronóstico de estos pacientes, por lo que es de importancia el conocimiento de esta entidad para su reconocimiento de forma precoz.




