







Los resultados obtenidos en investigación clínica con adultos no son, en muchas ocasiones, extrapolables a la población infantil. Además, hay enfermedades importantes de la infancia que son prácticamente incompatibles con la supervivencia en la edad adulta. Sin embargo, a pesar de las ventajas que pueda ofrecer determinada investigación clínica para verificar o comprobar la validez de un tratamiento, es potencial que cualquiera de los individuos (niños) pueda experimentar algún daño como consecuencia de participar en ella.
Con las garantías para la protección de grupos de población vulnerables que ofrece la legislación actual1,2 y los incentivos presentados por el Reglamento del Consejo Europeo, de 12 de diciembre de 20063, la investigación en pediatría está adquiriendo cada vez más desarrollo y protagonismo en la Comunidad Europea. Por tanto, es imprescindible que los jóvenes investigadores que afronten una investigación clínica en pediatría tengan conocimiento de las normas éticas y legales que rigen la investigación biomédica y sus especiales connotaciones. Asimismo, a la hora de confeccionar el diseño del protocolo, deben manejar las nociones básicas para diferenciar con precisión lo que se designa como proyectos de investigación y/o ensayos clínicos (EC). La excelencia investigadora, tanto científica como ética, es la salvaguarda más importante para la protección de los intereses y los derechos de los niños.
Puntos clave

La participación de los individuos en una investigación puede generar una situación de riesgo en la que fácilmente se pueden vulnerar sus derechos básicos. Por esta razón, debe conducirse de acuerdo con las normas éticas y legales que rigen la investigación en seres humanos4. La protección de grupos vulnerables, como la población pediátrica, ha sido causa de especial preocupación por parte de los agentes involucrados en investigación clínica.
Los comités éticos de investigación clínica (CEIC) tienen la misión de proteger la salud, la integridad y la dignidad de los individuos que van a someterse a investigación, en este caso particular de los menores. Deben evaluar los protocolos con sumo cuidado y, si fuera necesario, buscar asesoramiento en expertos con conocimiento profundo de aspectos clínicos, psicosociales y éticos pediátricos5. La minimización de molestias y riesgos es primordial en la investigación pediátrica.
El acuerdo tácito de los editores de revistas biomédicas de la Convención de Vancouver de no publicar los originales derivados de investigaciones clínicas que no haya aprobado un CEIC ha incrementado la necesidad de conocer los requerimientos en este sentido6.
Haciendo un breve recorrido histórico nos encontramos, en primer lugar, con el Código de Nüremberg (1947)7, que surge como consecuencia de la decisión judicial que condenó las atrocidades cometidas por el régimen nazi. Se centra en la necesidad de obtener el consentimiento del individuo y en el equilibrio favorable de la relación riesgo/beneficio. Sin embargo, no trata de la justa selección de los individuos, ni de la evaluación independiente de los ensayos. Posteriormente, la Asociación Médica Mundial desarrolla la Declaración de Helsinki (1964), con sus revisiones posteriores (Tokio 1975, Venecia 1993, Hong Kong 1989, Somerset West (Sudáfrica) 1996, Edimburgo 2000, con notas de clarificación al párrafo 29 en 2002 y al párrafo 30 en 2004, y Seúl 2008)8, que introduce por primera vez los principios éticos que debe seguir el médico para llevar a cabo una investigación con seres humanos y trata de distinguir entre la investigación médica asociada a la asistencia y la investigación biomédica no terapéutica con seres humanos. Establece como misión fundamental del médico proteger y salvaguardar la salud de los pacientes, incluidos los que van a someterse a investigación. Expresa claramente, como obligación de los médicos que participan en investigación, la protección de la dignidad, la integridad y los derechos a la autodeterminación, intimidad y confidencialidad de la información de los individuos de la investigación. En la práctica, esta distinción resulta muchas veces difícil de establecer, de la misma forma que es difícil delimitar la función médica asistencial de la investigación. El informe Belmont (1979)9 establece, por el contrario, una línea continua entre ambas actividades (asistencia-investigación) y establece unos principios básicos aplicables a todas las situaciones, centrándose en el consentimiento informado, en el equilibrio favorable de la relación riesgo/beneficio y en la necesidad de protección de las poblaciones vulnerables para que no sean objeto de investigaciones de riesgo. Las directrices éticas sobre investigación biomédica con seres humanos del CIOMS (Council for International Organizations of Medical Sciences) —propuestas en 1982, revisadas en 1993 y 1999, y publicadas finalmente en 2002— recogen, además, una sección sobre la compensación para los individuos en caso de presentar daños derivados de la investigación10. En la tabla 1 se recoge una selección de normas éticas fundamentales para la investigación biomédica en seres humanos.
Selección de normas éticas fundamentales para la investigación biomédica en seres humanos
| Año y revisiones | Fuente | |
|---|---|---|
| Código de Nüremberg | 1947 | Tribunal Militar de Nüremberg, decisión en los Estados Unidos |
| Declaración de Helsinki | 1964,1975, 1983,1989, 1996, 2000, 2002, 2004, 2008 | Asociación Médica Mundial |
| Informe Belmont | 1979 | Comisión nacional para la protección de los seres humanos en investigación biomédica y conductual |
| Guías éticas internacionales sobre investigación biomédica con seres humanos | Propuestas en 1982, revisadas en 1993, aprobadas en 2002 | Consejo para las organizaciones internacionales de ciencias médicas en colaboración con la Organización Mundial de la Salud |
| Las guías consolidadas de normas de buena práctica clínica | 1996 | Conferencia internacional para la armonización de los requisitos técnicos para el registro de medicamentos para uso humano |
| Convenio para la protección de los derechos humanos con respecto a las aplicaciones de la biología y la medicina. Convenio de Oviedo | 1997 | Consejo de Europa |
Las normas de buena práctica clínica (NBPC) surgieron de la necesidad de garantizar la calidad de la investigación y la protección de los derechos de los individuos en investigación clínica, con la finalidad de controlar el registro para la comercialización de nuevos medicamentos. Las estableció la Food and Drug Administration (FDA) de Estados Unidos en 1977 y posteriormente se implementaron en Europa, donde son de obligado cumplimiento desde 19911,11.
Se debe tener siempre presente que la población pediátrica está legalmente incapacitada para consentir y es dependiente de los adultos para su protección. El consentimiento informado deberá siempre obtenerse de los padres o representantes legales. Cuando los niños estén en condiciones de comprender la información, y siempre entre los 12 y 17 años, se deberá proporcionar una hoja de información adaptada a su grado de comprensión y obtener su asentimiento previamente a su inclusión en el estudio12.
Normativa de los ensayos clínicosEl Real Decreto 223/2004, de 6 de febrero (RCL 2004, 325)2, incorpora la Directiva 2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de 2001 (LCEur 2001, 1529)1, que trata de armonizar las legislaciones de los estados miembros de la Unión Europea sobre EC con medicamentos en seres humanos y exige el cumplimiento de las NBPC por parte de todos los agentes involucrados en la investigación.
El EC es por definición: “Toda investigación en seres humanos que tenga la intención de descubrir o confirmar los efectos clínicos, farmacológicos o farmacodinámicos, y/o detectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación o con productos sanitarios para determinar su eficacia y/o seguridad”. Considerando un medicamento en investigación: “Una forma farmacéutica de una sustancia activa o placebo que se investiga o se utiliza como referencia en un EC, incluidos los productos con autorización cuando se utilicen o combinen (en la formulación o en el envase) de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación no autorizada, o para obtener más información sobre un uso autorizado”2.
Aunque la mayoría de los EC los promueve la industria farmacéutica, los investigadores no deben dejar pasar oportunidades como la ofrecida en los años 2007 y 2008 por la Administración pública con las “Convocatorias de ayudas para proyectos de investigación clínica de carácter no comercial con medicamentos”. En el año 2008 se han presentado 304 presolicitudes, de las que 251 han sido seleccionadas y se encuentran en fase de evaluación. Desde la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) se están haciendo grandes esfuerzos para formar a los investigadores/ promotores independientes que cuentan con escasa capacidad de apoyo metodológico, administrativo y económico para poner en marcha estos ensayos (http://www.agemed.es/profHumana/ensayosClinicos/ayudas-IC.htm).
El documento “Aclaraciones sobre la aplicación de la Normativa de ensayos clínicos desde el 1 de mayo de 2004 (versión n.º 6, mayo de 2008)” contiene las instrucciones sobre cómo presentar una solicitud de EC a la AEMPS o a un CEIC.
Como regla general, podemos afirmar que todos los estudios en que se asigne a los pacientes un determinado tratamiento de forma aleatoria deben obtener la autorización de la AEMPS. El hecho de que todos los medicamentos se utilicen en el ensayo de acuerdo con las condiciones de uso autorizadas o habituales no exime de la autorización de la AEMPS, ni tampoco el que no todos los tratamientos que se comparen sean medicamentos (p. ej., cirugía frente a fármacos).
La aprobación del ensayo por parte de un CEIC es requisito imprescindible para la obtención de la autorización de la AEMPS.
Para estudios con otros diseños, debe tenerse en cuenta que se considera EC con medicamentos cualquier proyecto de investigación que estudie los efectos de un medicamento, a menos que cumpla todas las condiciones siguientes:
- 1.
El medicamento esté autorizado en España.
- 2.
Los medicamentos se prescriban de la manera habitual, de acuerdo con las condiciones autorizadas en ficha técnica.
- 3.
La asignación de los pacientes a una estrategia terapéutica no esté decidida de antemano por el protocolo, sino por la práctica clínica habitual.
- 4.
La decisión de prescribir un medicamento esté claramente disociada de la decisión de incluir al paciente en el estudio.
- 5.
No se aplique a los pacientes ninguna intervención, ya sea diagnóstica o de seguimiento, que no sea la práctica habitual.
Los estudios que se ajusten a las condiciones enumeradas, y cuando uno o varios medicamentos sean los factores de exposición fundamentales, deben remitirse a la AEMPS para su clasificación y registro como estudios postautorización, después de obtener la aprobación de un CEIC. La AEMPS informará a los investigadores promotores de los pasos que seguir.
Toda la legislación aplicable y otras guías de interés se encuentran disponibles en http://www.agemed.es/actividad/ invClinica/ensayosClinicos.htm
Normativa de las investigaciones biomédicasLa Ley 14/2007, de 3 de julio, de investigación biomédica13, abarca la investigación básica y la clínica, con exclusión de los EC con medicamentos y el implante de órganos, tejidos y células. En ella se recoge que los comités de ética de la investigación (CEI) deben garantizar la adecuación de los aspectos metodológicos, éticos y jurídicos de las investigaciones con intervención en seres humanos o la utilización de muestras biológicas de origen humano. Este papel fundamental de los CEI está siendo llevada a cabo por los actuales CEIC.
En España, las investigaciones biomédicas se han realizado en un conjunto muy variado de instituciones (universidades, centros de investigación, hospitales, fundaciones, laboratorios, etc.), entre las cuales cabe destacar el protagonismo de los centros universitarios y de los hospitales públicos en su desarrollo, gracias al apoyo prestado por el Fondo de Investigación Sanitaria y de otras entidades financiadoras14.
Los jóvenes investigadores no deben dejar pasar oportunidades como las propuestas por el Plan Nacional de I+D+I 2008-2011 que, en su Estrategia Nacional de Ciencia y Tecnología (ENCYT), publica convocatorias como las del Instituto de Salud Carlos III, de 15 de marzo de 2008, de concesión de ayudas a la Acción Estratégica en Salud, y cuyo objetivo es aumentar la inversión pública y privada en I+D+I en salud, potenciar la cantidad y la calidad de los recursos humanos en investigación en salud y aumentar la producción científica y la dimensión internacional de la investigación en salud mediante varias líneas de actuación15.
Como ya hemos explicado en el punto sobre la normativa de los EC, al diseñar un proyecto de investigación es imprescindible tener en cuenta que una investigación con intervención es aquella en la que se cambian las condiciones habituales de la práctica médica (aleatorización de tratamientos o medios diagnósticos y su enmascaramiento, selección de pacientes, comparación entre grupos, etc.). Por el contrario, un estudio observacional es aquel en el que no se modifican las condiciones habituales de la práctica clínica. La obtención de cualquier muestra biológica humana por razones distintas de las asistenciales puede hacer que los estudios se consideren intervencionistas.
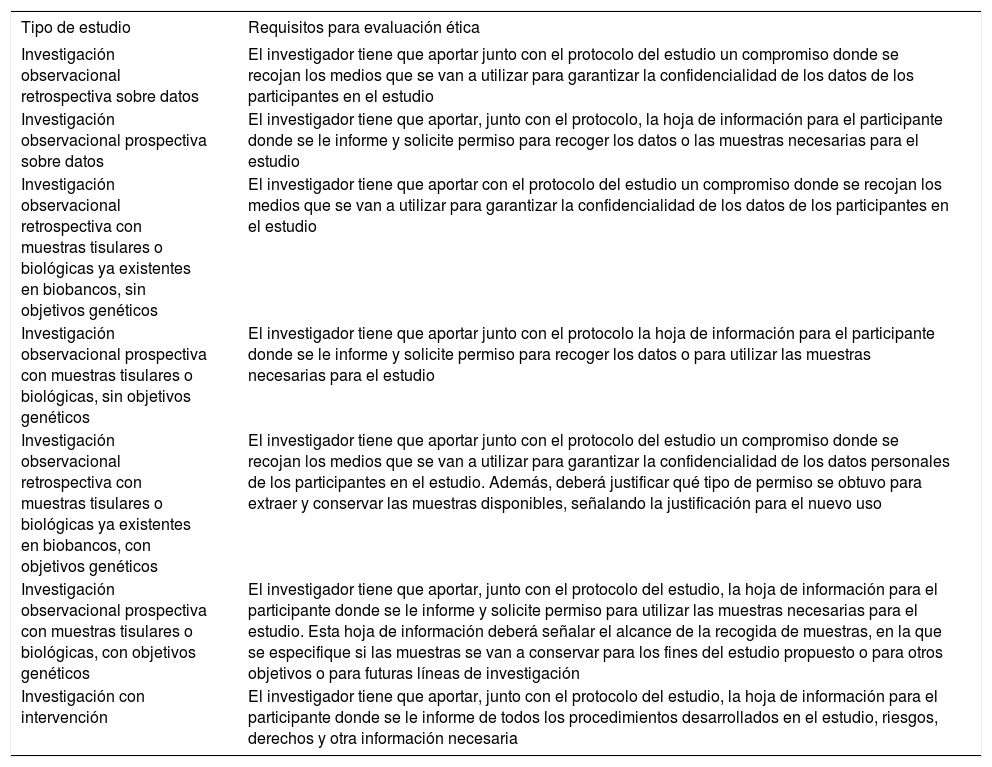
La tabla 2 ayudará a los investigadores a valorar el tipo de investigación que están protocolizando e informa de los requisitos necesarios para su presentación y solicitud de evaluación a los CEIC. La relevancia de la investigación en pediatría para nuestra sociedad científica y para la población pediátrica deben reconocerla, además, los responsables de nuestros hospitales y las actuales fundaciones de investigación, los cuales han de entender la investigación como tarea ineludible de los médicos asistenciales y que ello implica tiempo y recursos para llevarla a cabo16.
Clasificación de estudios y sus requisitos para evaluación ética
| Tipo de estudio | Requisitos para evaluación ética |
|---|---|
| Investigación observacional retrospectiva sobre datos | El investigador tiene que aportar junto con el protocolo del estudio un compromiso donde se recojan los medios que se van a utilizar para garantizar la confidencialidad de los datos de los participantes en el estudio |
| Investigación observacional prospectiva sobre datos | El investigador tiene que aportar, junto con el protocolo, la hoja de información para el participante donde se le informe y solicite permiso para recoger los datos o las muestras necesarias para el estudio |
| Investigación observacional retrospectiva con muestras tisulares o biológicas ya existentes en biobancos, sin objetivos genéticos | El investigador tiene que aportar con el protocolo del estudio un compromiso donde se recojan los medios que se van a utilizar para garantizar la confidencialidad de los datos de los participantes en el estudio |
| Investigación observacional prospectiva con muestras tisulares o biológicas, sin objetivos genéticos | El investigador tiene que aportar junto con el protocolo la hoja de información para el participante donde se le informe y solicite permiso para recoger los datos o para utilizar las muestras necesarias para el estudio |
| Investigación observacional retrospectiva con muestras tisulares o biológicas ya existentes en biobancos, con objetivos genéticos | El investigador tiene que aportar junto con el protocolo del estudio un compromiso donde se recojan los medios que se van a utilizar para garantizar la confidencialidad de los datos personales de los participantes en el estudio. Además, deberá justificar qué tipo de permiso se obtuvo para extraer y conservar las muestras disponibles, señalando la justificación para el nuevo uso |
| Investigación observacional prospectiva con muestras tisulares o biológicas, con objetivos genéticos | El investigador tiene que aportar, junto con el protocolo del estudio, la hoja de información para el participante donde se le informe y solicite permiso para utilizar las muestras necesarias para el estudio. Esta hoja de información deberá señalar el alcance de la recogida de muestras, en la que se especifique si las muestras se van a conservar para los fines del estudio propuesto o para otros objetivos o para futuras líneas de investigación |
| Investigación con intervención | El investigador tiene que aportar, junto con el protocolo del estudio, la hoja de información para el participante donde se le informe de todos los procedimientos desarrollados en el estudio, riesgos, derechos y otra información necesaria |


