





Puntos clave
- •
El síndrome nefrótico (SN) en el primer año de vida se comporta como una enfermedad monogénica donde la mutación de 4 genes explican la mayoría de los casos.
- •
Las mutaciones detectadas se relacionan con proteínas estructurales que forman parte de los elementos de la barrera de filtración glomerular, así como con proteínas reguladoras de los mismos.
- •
El comportamiento clínico es un SN corticorresistente, sin respuesta habitual a los regímenes esteroideos o de inmunosupresores.
- •
El diagnóstico etiológico se apoya en el estudio genético, identificando mutaciones en alguno de los genes conocidos, en función de los datos clínicos y la biopsia renal.
- •
La evolución a enfermedad renal terminal es la norma.
El tratamiento persigue el soporte clínico y nutricional del paciente, que conduzcan a unas condiciones adecuadas para el tratamiento renal sustitutivo.
- •
Es importante un adecuado consejo genético a las familias con casos conocidos o nuevos, de cara a la planificación de la descendencia.
Lectura rápida
El síndrome nefrótico (SN) es la glomerulopatía más frecuente en la infancia. Está caracterizado por la pérdida masiva de proteínas en la orina (> 40mg/m2/h), con la presencia de hipoalbuminemia (< 2,5g/dl) y clínica de edemas, así como hiperlipidemia. Su presentación más habitual es en la edad escolar, donde suele ser idiopático.
Una proporción muy elevada de los casos de SN que comienzan en el primer año de vida tienen como base causal una alteración genética. Estos desórdenes no son, sin embargo, un hecho exclusivo de este grupo de edad, ya que explican un parte de aquellos SN que se desarrollan en edades superiores, en situación de resistencia a los tratamientos convencionales.
Las mutaciones están referidas principalmente a genes que codifican para proteínas del podocito, que es una parte esencial de la barrera de filtración glomerular. Estas alteraciones conllevan una ausencia de producción de la proteína o bien un menor actividad en su función. Este hecho implica una alteración estructural, a diferencia de lo que sucede en el SN idiopático, con amplia base inmunológica.
El SN congénito (SNC) es aquel presente antes del tercer mes de vida, incluso intraútero. Los genes más frecuentemente implicados son NPHS1 (que codifica para nefrina, una de las proteínas del diafragma de filtración, que se describe sobre todo en la población finlandesa). El otro gen más relacionado es NPHS2 (codifica para la podocina; otra de las proteínas de membrana del podocito, muy frecuentemente descrita fuera de esta población nórdica).
El SN infantil (SNIn) es aquel que se presenta entre el 4.° y el 12.° mes de vida. La mutación más frecuentemente encontrada es la que afecta al gen NPHS2.
Existen casos de SN en el primer año de vida en los que no se identifica mutación. Actualmente, se conocen gran variedad de genes implicados en el mismo, si bien en el 66% de los casos se corresponde con mutaciones en los genes NPHS1, NPHS2, WT1 o PLCε1.
El curso clínico de estos pacientes depende de la mutación encontrada. Existe una amplia heterogeneidad fenotípica para mutaciones del mismo gen y para pacientes con similar mutación hallada. Existen mutaciones que conllevan un espectro clínico más leve, con respuesta en ocasiones al tratamiento corticoideo o inmunosupresor estándar.
El estudio genético es una parte clave en el diagnóstico de estos pacientes. Sin embargo, debe ser organizado y, en su caso, en varios pasos. La selección y el orden de los genes que deben secuenciarse deben hacerse considerando la edad del comienzo del SN, la asociación de otros síntomas en un síndrome y la histología obtenida de la biopsia renal.
Al ser una enfermedad genética y heredable, es importante el consejo genético a las familias a la hora de la planificación de su descendencia. Esta circunstancia debe contemplarse tanto en el caso de que haya un miembro afecto de SN, como si se planea la misma desde un estado de portador.
En la mayoría de los casos de SN en el primer año de vida no existe respuesta a los tratamientos convencionales (corticorresistencia), lo que se sigue de una progresión hacia la enfermedad renal crónica (ERC). Esta evolución es variable en el tiempo, oscilando desde unos pocos meses hasta la edad adulta, en función del tipo de gen alterado y la mutación concreta.
El manejo de estos pacientes está dirigido hacia el soporte clínico de los mismos, para control de los edemas, evitar las infecciones y conseguir una nutrición adecuada. De esta manera, se consigue un estado óptimo en su evolución hacia la ERC.
El tratamiento, en la mayoría de los casos, pasa por el tratamiento renal sustitutivo. Existen diferentes vías, que tienen como nexo final el trasplante renal, habitualmente de donante cadáver. Si bien es cierto que en la mayoría de los casos se habla de un defecto estructural, no es esperable que se reproduzca la proteinuria posteriormente, pero se han descrito casos de recurrencia. En general, el pronóstico del paciente y del injerto es similar al de otras causas de trasplante renal.
El síndrome nefrótico (SN) es un desorden que se caracteriza por la pérdida masiva de proteínas en la orina (> 40mg/m2/h), hipoalbuminemia (< 2,5g/dl) y edemas1. La presentación habitual es la etapa escolar (2-8 años). En estos casos, la causa suele ser desconocida, denominándose síndrome nefrótico idiopático (SNId). El tratamiento tiene como base los corticoides y, en ocasiones, precisa combinar varios fármacos inmunosupresores. Se suele obtener curación tras diferentes recaídas en su historia natural.
El SN en el primer año de vida (SNPA) tiene otras características. Cuando se desarrolla en los primeros 3 meses se llama SN congénito (SNC) y entre los 4-12 meses, síndrome nefrótico infantil (SNIn). Tiene una fuerte base genética, que produce un defecto estructural en la barrera de filtración o en proteínas reguladoras. Esta clasificación ayuda al diagnóstico en la clínica, pero las alteraciones genéticas que se presentan se pueden expresar en diferentes edades2.
EtiologíaEn la tabla 1 se relacionan las causas que se han relacionado con el SNPA. Las formas primarias se han asociado a alteraciones en algún gen, además de las idiopáticas. Los SN asociados a un cortejo sintomático se agrupan como sindrómicos.
Principales causas de síndrome nefrótico en el primer año de vida. Clasificación clínica.
| Primario |
| Síndrome nefrótico congénito |
| Mutación en el gen de la nefrina (NPHS1). Tipo |
| finlandés |
| Idiopático |
| Síndrome nefrótico infantil |
| Mutación del gen de la podocina (NPHS2) |
| Mutación gen PLCε1 |
| Idiopático |
| Sindrómicos |
| Mutación gen WT1. Síndrome de Denys-Drash. |
| Frasier |
| Mutación gen LAMB2. Síndrome de Pierson |
| Mutación gen LMX1B. Síndrome de nail-patella |
| Mutación gen LAMB3. Epidermólisis ampollosa de Herlitz |
| Otros |
| Síndrome de Galloway-Mowat |
| Miopatías mitocondriales |
| SN con o sin malformaciones cerebrales y otras (no defecto genético identificado) |
| Secundario |
| TORCHS: Sífilis, toxoplasmosis, CMV, rubéola |
| Hepatitis B |
| Malaria |
| VIH |
| Lupus eritematoso sistémico materno |
| Autoanticuerpos maternos contra la endopeptidasa neutral neonatal |
| Tratamiento materno con clorfeniramina |
| Exposición a mercurio |
CMV: citomegalovirus; SN: síndrome nefrótico; VIH: virus de la inmunodeficiencia humana.
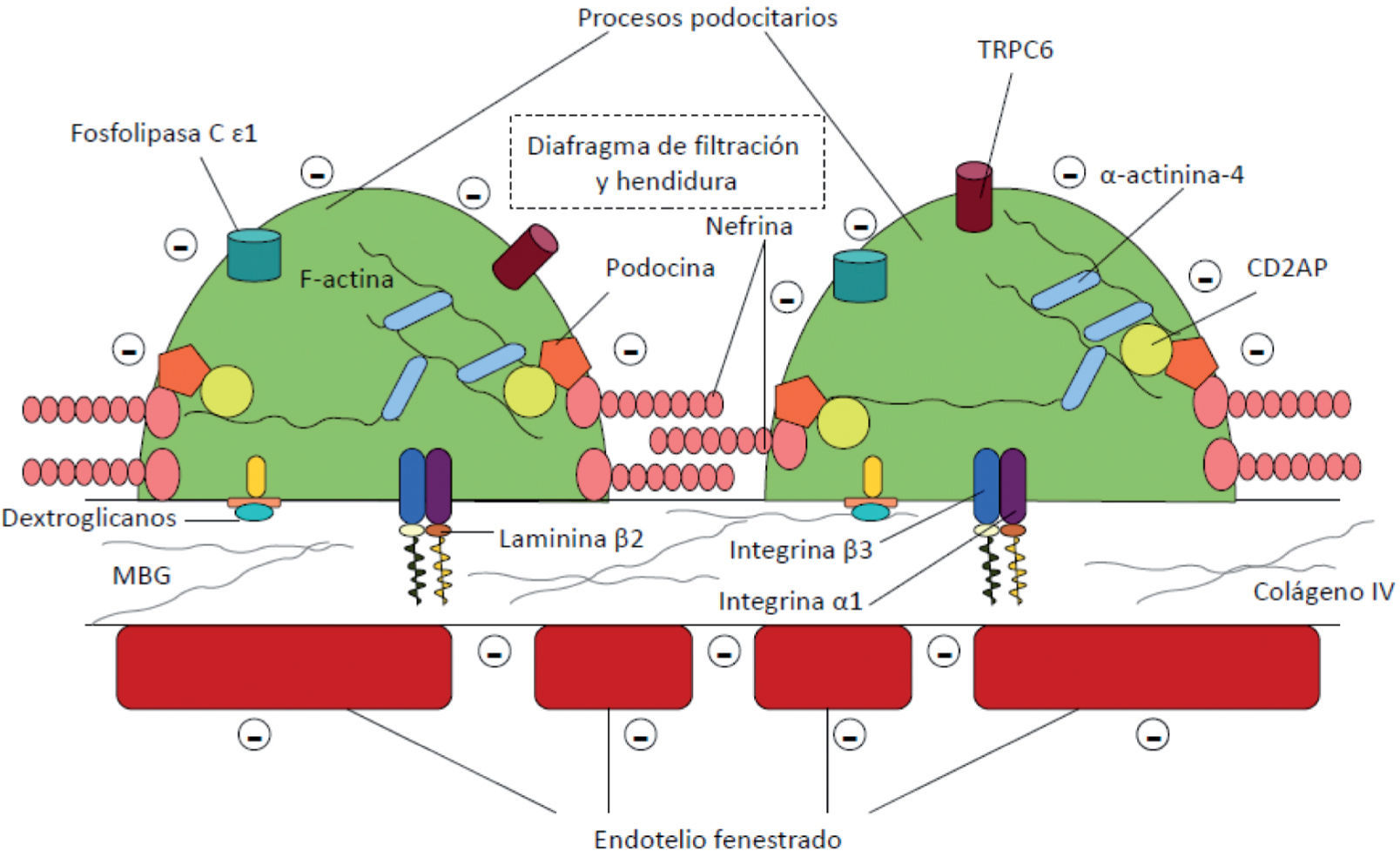
El elemento clave del SN es la pérdida masiva de proteínas a la orina por paso a través de la barrera de filtración glomerular (BFG). Esta está compuesta por 3 elementos (fig. 1):
. MBG: membrana basal glomerular.")
- –
Endotelio fenestrado capilar.
- –
Membrana basal glomerular (MBG).
- –
Epitelio celular (podocitos) que presentan distalmente proyecciones digitiformes que generan espacios entre ellos (hendidura de filtración).
Esta barrera actúa como un filtro eficaz para las moléculas, en razón de su carga y tamaño, y solo el agua y las sustancias plasmáticas pequeñas se filtran al espacio de Bowman. Por ello, la alteración de alguno de estos elementos puede conducir al filtrado anómalo de sustancias a la orina. De los 3 elementos, el podocito y sus alteraciones desempeñan el papel principal en la génesis de la proteinuria3. La MBG es un esqueleto en el que predomina el colágeno tipo IV, laminina (confiere soporte a la MBG), enactina, nidogeno (ambas permiten el anclaje del colágeno y laminina) y proteoglucanos cargados negativamente (parecen tener un papel activo en la permeabilidad de sustancias cargadas negativamente; hoy está más cuestionado este hecho)4,5.
El podocito es una célula altamente diferenciada que proviene de células mesenquimales3,5. El cuerpo celular está en el centro de la misma, con las organelas citoplasmáticas y el núcleo, y se proyecta sobre el espacio urinario. Del cuerpo celular se originan los procesos primarios, que son estructuras alargadas que contienen los procesos podocitarios. Estos se relacionan y se anclan con la MBG a través de proteínas de la familia de las integrinas y dextroglucanos. Los procesos podocitarios se entrelazan entre sí a modo de interdigitación con espacios de 40nm. Están cubiertos por una membrana extracelular (constituida principalmente por nefrina), formando el diafragma de hendidura. La estructura podocitaria es compleja y en su mantenimiento desempeñan un papel muy importante la actina (auténtico esqueleto de la célula) y las proteínas que interaccionan con ella (sinaptopodina y α-actinina-4). Esta configuración le permite un dinamismo para cambiar de forma. A través de esta estructura, se consiguen los filtros de tamaño y carga, el mantenimiento de la estructura del ovillo capilar, contrarrestar la presión intraglomerular, la síntesis y mantenimiento de la MBG y la producción del factor de crecimiento vascular endotelial (necesario para la integridad el endotelio fenestrado)6. Merced a proteínas, se conecta la hendidura de filtración con el esqueleto de actina y colaboran en la transducción de señales en el podocito. La pérdida de estructura molecular conduce la retracción del podocito (en inglés, effacement). La hendidura, junto con la capacidad dinámica de los procesos podocitarios, son los elementos centrales de la barrera de filtración3,5.
Las proteínas involucradas en este tamiz y sus elementos tienen codificación en genes para los que se han identificado mutaciones relacionadas con el SN en el primer año de vida y otros desórdenes proteinúricos2,6.
Síndrome nefrótico y corticorresistenciaEl tratamiento del SN son los corticoides, debido a que patogénicamente se ha sugerido una alteración inmunitaria (un factor proteinúrico no identificado que parece provenir de una disfunción de células T). Si la respuesta a los mismos es favorable, se clasifican como SN corticosensibles (SNCS), y si no se produce, SN corticorresistentes (SNCR).
El 90% de los SNId son SNCS. Los restantes son SNCR y desarrollan enfermedad renal crónica (ERC) en un 30-40%, tras 10 años de seguimiento7. Ante una situación de corticorresistencia, está indicada la realización de biopsia renal8.
Por oposición, estudios recientes destacan que alteraciones estructurales en la barrera de filtración glomerular causan una importante proporción de casos de SNCR.
Dentro del concepto de corticorresistencia (CR), existen formas asociadas a otras anomalías (formas sindrómicas) y otras que son alteraciones aisladas. Dentro de las formas no sindrómicas, se han encontrado alteraciones en los genes NPHS1, NPHS2, CD2AP, PLCε1, ACTN4, TRPC6 e INF2.
Las formas sindrómicas son menos frecuentes y se deben a alteraciones en genes que codifican para factores de transcripción (WT1, LMX1B), elementos de la MBG (LAMB2, ITGB4), proteínas lisosomales (SCARB2), mitocondriales (COQ2, PDSS2, MTTL1) o un mediador de reestructuración del ADN-nucleosoma (SMARCAL1). Presentan heterogeneidad clínica, ya que las mutaciones en estos genes no siempre se acompañan de asociación sindrómica, como es el caso de WT1 y LAMB27.
El aumento de los genes identificados responsables en su alteración de SN está aumentando. Así, el estudio genético es cada vez una tarea más compleja que precisa de información clínica e histológica, de cara a orientar el estudio de genes. Si bien es cierto que suele existir una alteración genética, hasta en un 18% de los casos de SNC, y un 33% dentro del primer año, no se encuentra alteración en los genes estudiados y conocidos9,10.
Correlación genéticoclínicaExisten numerosas publicaciones acerca de las mutaciones debidas a NPHS1, en el llamado SN finlandés (SNF). Globalmente, el 66% de los casos de SNPA se explican por la mutación en 4 genes (NPHS1, NPHS2, WT1, PLCε1), por lo que, de forma importante, el SNPA es una enfermedad monogénica10.
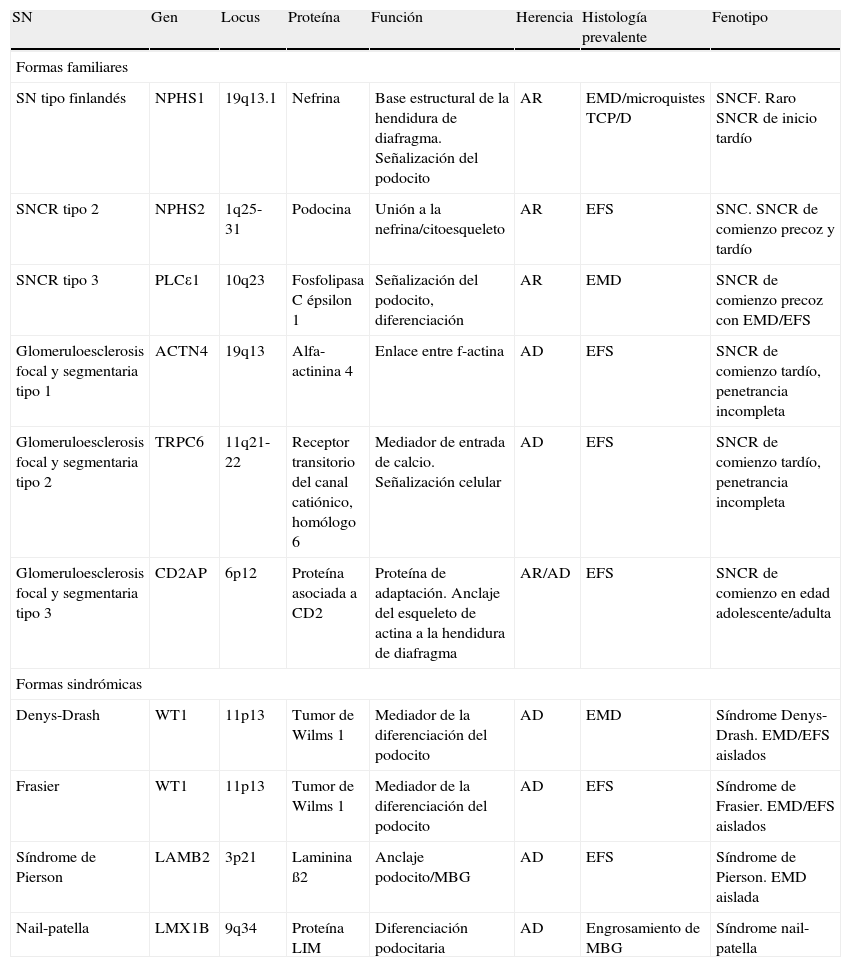
La mutación más habitualmente encontrada responsable de SNC es la de NPHS1 en población finlandesa, de la misma forma que globalmente mutaciones en NPHS2 son las más descritas en el SNCR de inicio precoz7. En la tabla 2 se resumen los genes implicados más importantes, así como sus características.
Principales genes involucrados en SN familiares y en formas sindrómicas.
| SN | Gen | Locus | Proteína | Función | Herencia | Histología prevalente | Fenotipo |
| Formas familiares | |||||||
| SN tipo finlandés | NPHS1 | 19q13.1 | Nefrina | Base estructural de la hendidura de diafragma. Señalización del podocito | AR | EMD/microquistes TCP/D | SNCF. Raro SNCR de inicio tardío |
| SNCR tipo 2 | NPHS2 | 1q25-31 | Podocina | Unión a la nefrina/citoesqueleto | AR | EFS | SNC. SNCR de comienzo precoz y tardío |
| SNCR tipo 3 | PLCε1 | 10q23 | Fosfolipasa C épsilon 1 | Señalización del podocito, diferenciación | AR | EMD | SNCR de comienzo precoz con EMD/EFS |
| Glomeruloesclerosis focal y segmentaria tipo 1 | ACTN4 | 19q13 | Alfa-actinina 4 | Enlace entre f-actina | AD | EFS | SNCR de comienzo tardío, penetrancia incompleta |
| Glomeruloesclerosis focal y segmentaria tipo 2 | TRPC6 | 11q21-22 | Receptor transitorio del canal catiónico, homólogo 6 | Mediador de entrada de calcio. Señalización celular | AD | EFS | SNCR de comienzo tardío, penetrancia incompleta |
| Glomeruloesclerosis focal y segmentaria tipo 3 | CD2AP | 6p12 | Proteína asociada a CD2 | Proteína de adaptación. Anclaje del esqueleto de actina a la hendidura de diafragma | AR/AD | EFS | SNCR de comienzo en edad adolescente/adulta |
| Formas sindrómicas | |||||||
| Denys-Drash | WT1 | 11p13 | Tumor de Wilms 1 | Mediador de la diferenciación del podocito | AD | EMD | Síndrome Denys-Drash. EMD/EFS aislados |
| Frasier | WT1 | 11p13 | Tumor de Wilms 1 | Mediador de la diferenciación del podocito | AD | EFS | Síndrome de Frasier. EMD/EFS aislados |
| Síndrome de Pierson | LAMB2 | 3p21 | Laminina ß2 | Anclaje podocito/MBG | AD | EFS | Síndrome de Pierson. EMD aislada |
| Nail-patella | LMX1B | 9q34 | Proteína LIM | Diferenciación podocitaria | AD | Engrosamiento de MBG | Síndrome nail-patella |
AD: autosómico dominante; AR: autosómico recesivo; EMD: esclerosis mesangial difusa; EFS: esclerosis focal y segmentaria; MBG: membrana basal glomerular; SN: síndrome nefrótico; SNCR: síndrome nefrótico corticorresistente; SNFC: síndrome nefrótico tipo finlandés; TCP/D: túbulo contorneado proximal/distal.
Es la forma más frecuente de SNC y es más habitual en Finlandia, donde tiene una incidencia de 1 caso cada 8.200 recién nacidos (RN) vivos7. Es un trastorno autosómico recesivo por mutaciones en el gen de NPHS1, que codifica para la nefrina, proteína de adhesión transmembrana de la superfamilia de las inmunoglobulinas2,6,7. Su síntesis es casi exclusiva en los podocitos y es un componente crucial del diafragma de hendidura. Las mutaciones en NPHS1 se traducen en alteraciones en tráfico intracelular de la proteína, así como en su función. Se han documentado más de 140 mutaciones diferentes. Las principales se denominan Fin-major (exón 2, p.L41fsX91) y Fin-minor (exón 26, p.R1109X), que se detectan en el 78 y el 16% de los alelos mutados entre los pacientes finlandeses7,11. Entre la población no finlandesa, la mutación en NPHS1 es inferior, el 39-55% de los casos con perfil clínico de SNF7,9.
Presenta menos variabilidad fenotípica que otros SN genéticos. La mayoría de estos pacientes nacen de forma prematura, con pesos entre los 1.500-3.000g. La placenta está aumentada de tamaño (hasta el 25% del peso del recién nacido). No se han descrito malformaciones extrarrenales, pero sí alteraciones funcionales menores (hipotonía e hipertrofia cardiaca)3. Los estudios demuestran que las alteraciones en NPHS1 comienzan con más precocidad que las de NPHS29. Clínicamente, se caracteriza por una proteinuria masiva al nacimiento (iniciada ya intraútero y detectable en la primera orina del recién nacido), así como hematuria microscópica y creatinina normal en los primeros meses. Sin embargo, no todas las mutaciones del NPHS1 se asocian a enfermedad grave o evolución complicada4. El curso de la enfermedad es rápido, con evolución habitual hacia ERC en la primera década de la vida6.
La anatomía patológica es variable, con hallazgos de expansión mesangial y dilataciones radiales de los túbulos proximales y distales (aunque inconstantes)2,7. Con el tiempo y la evolución, se pueden ver fibrosis intersticial e infiltrados inflamatorios, sobre todo en los glomérulos.
Mutaciones en NPHS2 (OMIM#604766)El gen NPHS2 codifica para la podocina, proteína podocitaria sobre la que cada vez existe más conocimiento acerca de su importancia en la BFG. Permite la correcta ubicación de la nefrina en el diafragma de hendidura, interaccionando además con la proteína CD2AP. Su expresión está restringida al podocito.
Su herencia sigue un patrón autosómico recesivo. Sus mutaciones se han detectado hasta en el 51% de casos de SNC en pacientes centroeuropeos2,5 y, como SNIn, hasta el 35% de los pacientes10. Del global de SNCR, su mutación ocurre en un 40% como familiar y un 6-17% de casos esporádicos6,7. Se han descrito más de 100 alteraciones patogénicas3,7.
Las alteraciones ligadas a NPHS2 se describieron inicialmente como SNCR infantil, pero posteriormente se han encontrado casos desde el nacimiento hasta la edad adulta3. Presenta más variabilidad fenotípica que las debidas a NPHS1, con espectro clínico más leve. No hay asociación de alteraciones extrarrenales, aunque sí se han descrito problemas cardiacos menores2.
La anatomía patológica suele presentar esclerosis focal y segmentaria (EFS), si bien las biopsias precoces suelen revelar cambios mínimos glomerulares. El desarrollo de ERC se produce habitualmente en la primera década de la vida2,7. Sin embargo, se describen mutaciones con comienzo más tardío y fenotipo clínico más leve, y evolución hacia ERC a edades superiores9.
Síndrome nefrótico infantilEn este grupo, la mutación de NPHS2 es la más habitual. Recientemente, se ha descrito que las mutaciones en NPHS1 también son responsables de una proporción de casos de SNIn y de SNCR de instauración por encima de los 3 meses7.
Otros genes implicados en el desarrollo de SNCR de comienzo en la edad infantil son PLCε1 y WT1, cuya histología habitual es la esclerosis mesangial difusa (EMD). Esta anatomía patológica se encuentra en otras formas sindrómicas (Pierson, Galloway- Mowat). Sin embargo, el gen más frecuentemente implicado en esta histología son mutaciones en PCLε17.
Mutaciones en PLCε1 (OMIM#609414)Este gen codifica para la proteína fosfolipasa C épsilon 1, enzima citoplasmática precisa para la maduración del podocito (hidroliza fosfolípidos de membrana para generar segundos mensajeros de rutas metabólicas intracelulares relacionadas con la diferenciación celular) y participa en mecanismos de adhesión celular.
Sus mutaciones suponen la principal causa de la histología de EMD (28,6% de familias con EMD6,7). Presenta un espectro clínico variado en función del tipo de mutación, que abarca desde el comienzo en los primeros días de vida hasta los 8 años7. Ocasionalmente, se describe histología asociada de EFS, con un perfil clínico más leve.
Mutaciones en WT1 (OMIM#607102)Las mutaciones en WT1 representan hasta el 9% de los SNCR no familiares6,7. Su herencia es autosómica dominante, pero existen formas familiares con herencia recesiva3. Es un gen supresor de tumores y codifica para un factor de regulación de la expresión de varios genes en el desarrollo de los riñones y resto de estructuras urológicas y genitales. Se expresa abundantemente en los podocitos y controla, entre otros, la expresión de nefrina. Su mutación se ha relacionado también con el síndrome de Denys-Drash, Frasier y esclerosis mesangial difusa con SN aislado.
La histología se ha documentado en casos de EMD y también en casos aislados de EFS.
Síndrome de Denys-DrashSNCR de instauración en los primeros meses, seudohermafroditismo masculino, disgenesia gonadal y tumor de Wilms hasta en el 90%3. La evolución hacia ERC es rápida. Su histología habitual es la EMD.
Síndrome de FrasierSNCR con seudohemarfroditismo masculino. Respecto a síndrome de Denys-Drash (SDD), las mutaciones son diferentes, la instauración de la proteinuria es más tardía y presenta una histología con EFS de progresión lenta hacia ERC hacia la 2.a-3.a década de la vida3. Pueden desarrollar gonadoblastoma.
Con todo, se describen mutaciones de síndrome de Frasier (FS) halladas en casos de SDD o EMD aislada, así como mutaciones típicas de SDD con histología de EFS o tumor de Wilms sin SN3 (9). También hay casos no sindrómicos con mutación aislada en el riñón, con debut como SNC2 (1).
Otras formas sindrómicasLAMB2 (OMIM#150325)Codifica para la proteína laminina-β2, que pertenece a la estructura de la MBG (además de la retina) y actúa de anclaje del podocito a la misma. Su alteración se traduce en el síndrome de Pierson, un SN de comienzo como SNC, que asocia microcoria (pero hay casos sin afectación ocular)3,12. La evolución es hacia una rápida ERC, con un patrón histológico de EMD.
Galloway-Mowat (OMIM#251300)Podocitos y neuronas comparten similitudes en proteínas estructurales y se describen casos de afectación combinada. Asocia SN, de debut precoz y alteraciones del SNC (retraso mental, microcefalia, alteraciones cerebrales). También se han descrito hernia de hiato, rasgos dismórficos, talla baja y defectos diafragmáticos. Su herencia es recesiva, pero se desconoce el gen/es implicado/s. La anatomía patológica no es característica y se describen patrones diferentes2,11.
LMX1B (OMIM#602575)Codifica para un factor de transcripción importante en el desarrollo precoz del podocito. Produce el síndrome nail-patella, trastorno autosómico dominante, con uñas displásicas y ausencia/displasia de patela, así como alteraciones glomerulares debidas a borramiento de los procesos podocitarios y engrosamiento de la MBG.
Otros genes descritosMutaciones en los genes CD2AP (OMIM#604241), ACTN4 (OMIM#604638), TRPC6 (OMIM# 6 0 3 6 5 2) e INF2 (OMIM#610982) (codifica para una familia de proteínas reguladoras de la polimerización y despolimerización de la actina del citoesqueleto) no han sido referidos en el SNPA; el SN asociado se desarrolla en la edad adulta y sigue un patrón de herencia autosómica dominante. Histológicamente presentan EFS7,10.
Formas no genéticas. Casos secundariosLa mayoría de los casos son infecciosos.
- 1.
Infecciones:
- –
Sífilis congénita. El SN es raro, y su curso más leve. El tratamiento con penicilina es curativo.
- –
Hepatitis B, toxoplasmosis, rubéola.
- –
Citomegalovirus: su detección en fase precoz puede producir confusión con un SN de base genética. Una mala respuesta a ganciclovir pone en la pista de este hecho.
- –
Infección por virus de la inmunodeficiencia humana: más frecuente tras el primer año de vida.
- –
- 2.
Lupus eritematoso sistémico materno.
- 3.
Autoanticuerpos maternos contra la endopeptidasa neutral neonatal (presente en los podocitos).
Existe amplia variabilidad de presentación, con casos que comienzan inmediatamente al nacimiento, y otros en las primeras semanas de vida. Las formas graves se presentan como edemas generalizados con excreción urinaria de proteínas aumentada y albúmina sérica baja. Formas menos sintomáticas pueden hacer más complicado el diagnóstico. Las cifras de creatinina suelen ser normales (sobre todo en mutaciones de NPHS1) pero en otras formas genéticas con daño renal establecido se eleva. La presión arterial oscila según sean el estado de la volemia y la presencia de fallo renal. La presencia de un cortejo sindrómico (afectación extrarrenal) ayuda a orientar el caso. La biopsia renal no esclarece la causa etiológica, ya que los diferentes tipos genéticos pueden producir varios tipos de afectación. Está indicada en el SNPA, ya que ayuda a orientar el estudio genético y decisiones de terapia farmacológica y sustitutiva.
Actualmente, el diagnóstico genético es de elección para el SNPA. Permite establecer el pronóstico de la enfermedad, el seguimiento y consejo genético.
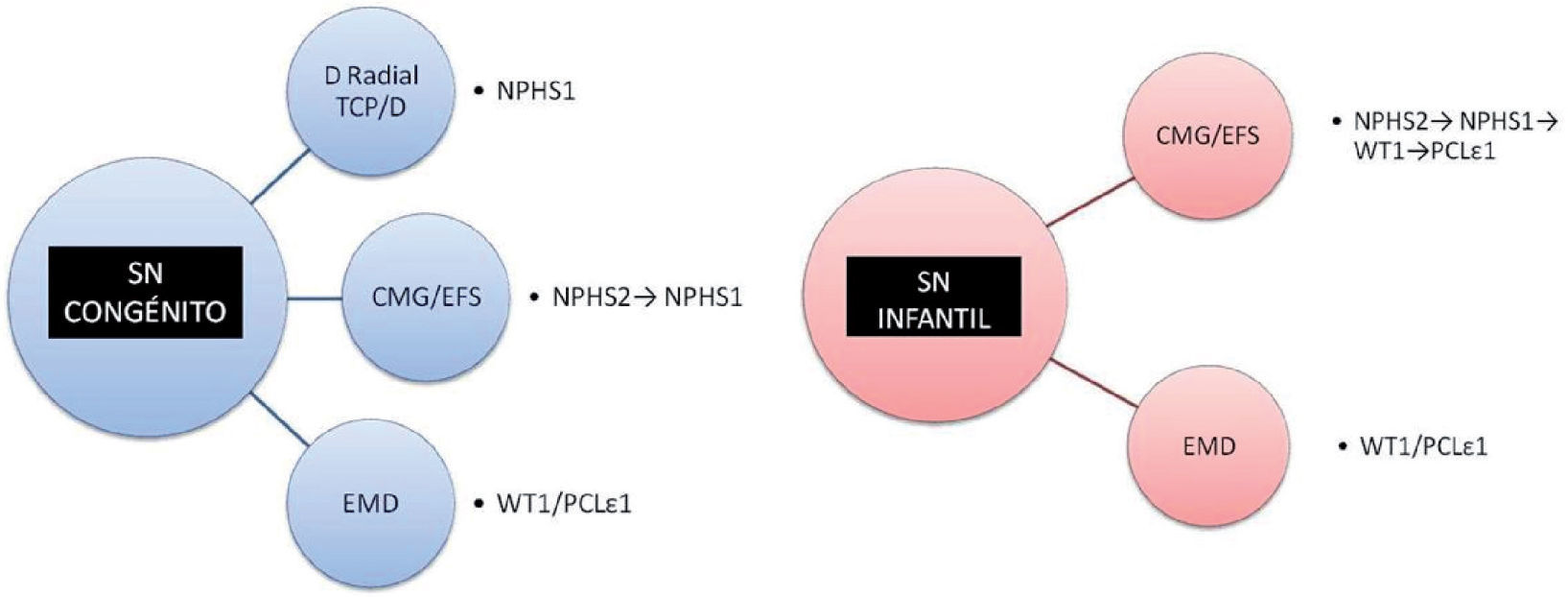
Estudio genéticoEn los casos de SNC no sindrómicos, el primer gen que se debe secuenciar es NPHS1, sobre todo si el comienzo se produce en los primeros días de vida y la histología presenta dilataciones tubulares proximales. En caso de no encontrarse mutaciones, se secuenciará NPHS2. Pacientes con comienzo más tardío (habitualmente tras 4 semanas de vida), con histología de mínimos cambios o EFS se debería buscar anomalías en NPHS2 seguido de NPHS1 en caso de que no se detecten. Para el resto de los pacientes en los que no se identifique mutaciones en estos genes, y con histología de EMD, se debería continuar con WT1/PCLε1.
En los casos de SNIn que se presentan como SNCR no sindrómicos, con histología de mínimos cambios o EFS, el primer gen a secuenciar sería NPHS2, seguido en su caso de NPHS1. Para los demás casos con esta histología, el siguiente gen a secuenciar sería WT1, siendo muy baja la probabilidad de hallar mutaciones de PCLε1 en este grupo (más rentable si es familiar). Sin embargo, en casos con histología de EMD de forma aislada, las mutaciones en PCLε1 son las más habitualmente encontradas, siendo sin embargo las mutaciones de WT1 en ocasiones priorizadas a este, por los altos costes en la determinación de PCLε17 (fig. 2).
Tratamiento
El tratamiento del SNPA es complejo, dada la ausencia de respuesta a tratamiento esteroideo e inmunosupresor (aunque se refieren respuestas parciales en mutaciones de WT1 a ciclosporina A4,10). Estos pacientes suelen requerir estancias iniciales prolongadas, con múltiples reingresos por infecciones y alteraciones hidroelectrolíticas. La ausencia de respuesta a las terapias convencionales puede sobrellevar una carga de efectos secundarios indeseables. Por ello, una vez obtenido un estudio genético concluyente, se debe suspender cualquier tratamiento inmunomodulador10.
El objetivo de la terapia durante los primeros meses persigue el control de los edemas y la uremia, prevención y tratamiento de complicaciones (infecciones y trombosis) y soporte nutricional. En muchos casos, la terapia renal sustitutiva con trasplante es la única curación3,13.
Infusiones de albúminaLas reposiciones no son curativas, pero controla los edemas así como la malnutrición y crecimiento. Se emplean infusiones de albúmina 20% a través de vía central (3-4g/kg/día), seguidas de bolo de furosemida. La proteinuria es menos grave en los casos con histologías de EMD, pudiendo no precisar terapia suplementaria11.
NutriciónLos aportes energéticos ascienden a unas 130Kcal/día, con proteínas en dosis de 3-4g/día. Se emplean fórmula artificial y lactancia materna. Precisan aportes de vitamina D y complejos vitamínicos habituales según la edad. Se deben corregir déficits de calcio y magnesio. Puede ser preciso el uso de sonda nasogástrica para conseguir estos aportes.
MedicacionesEl uso de inhibidores de la enzima conversora de angiotensina (IECA) se ha demostrado útil en algunos casos de SNPA reduciendo la proteinuria, en los que no existe una ausencia total de expresión de nefrina o podocina3,13. Las pérdidas urinarias de proteínas hacen que se suplementen con hormonas tiroideas y, por riesgo trombótico por pérdida de factores de la coagulación, se usa aspirina o warfarina. La pérdida de inmunoglobulinas predispone a una mayor tasa de infección Se recomienda el uso de antibióticos por vía parenteral en sospecha de infección, no profilácticos3.
Terapia sustitutiva: nefrectomía y trasplanteAlgunos centros realizan de rutina una nefrectomía unilateral precoz para control de proteinuria, con retraso del trasplante a edad más tardía3,14. En otros centros se realiza nefrectomía bilateral precoz con diálisis peritoneal, con trasplante de injerto con un peso de unos 9-10kg15. Un tercer manejo se ha contemplado, con trasplante renal precoz con nefrectomía en el mismo acto3. La mayoría de los pacientes se trasplantan a los 1-2 años de vida. La supervivencia a 5 años es en el paciente del 90% y del injerto del 80%3,6. Sin embargo, son pacientes que precisan retrasplante en la edad adulta por fallo crónico del mismo.
Recurrencia de la proteinuria postransplanteAunque para la situación de una alteración en la barrera de filtración, el trasplante no debería conllevar recurrencia de la proteinuria, se han descrito en pacientes portadores de mutaciones en NPHS1, NPHS2, ACTN4. En muchos casos, se desconoce la causa; en otros, se ha relacionado con anticuerpos dirigidos contra proteínas indemnes en el injerto7. También es importante la iatrogenia asociada a la inmunosupresión empleada en el mantenimiento del trasplante. Hasta la fecha, no se han descrito casos de recurrencia de la enfermedad postrasplante en mutaciones de WT14.
La donación de paciente vivo, en la que alguno de los progenitores es portador en heterocigosis y sano, debe ser cuidadosamente valorada previa al trasplante, debido a la incertidumbre de un eventual desarrollo de SN en donante y/o receptor7.
Consejo genético y diagnóstico prenatalSiempre que exista una alteración heredable en un paciente, se debe ofrecer un consejo genético a la hora de planificar la descendencia, contactando con genetistas.
En el caso de mutaciones en WT1, aunque la mayoría de las mutaciones se producen de novo, hay que plantear el escenario en el caso de mujeres XX con mutaciones en WT1 que conllevan EMD/EFS aislada. Estas tienen un desarrollo genital normal, pero pueden quedarse embarazadas y transmitir el gen mutado a su descendencia. El fenotipo depende del cariotipo: 46XY pueden desarrollar SDD o de FS; los 46XX no presentarán genitales ambiguos. En este caso, mujeres portadoras de mutaciones en WT1 que quieran quedarse embarazas han de ser advertidas de que su descendencia puede tener un fenotipo diferente del suyo.
En el caso de mutaciones de PCLε1, se debe advertir de la variabilidad del fenotipo renal. Esto es debido a que se han descrito casos en adultos homocigotos en la mutación de este gen que permanecen asintomáticos, con descendencia afecta con EMD. Probablemente, existen además para este gen otros que actúen como modificadores, y otros factores ambientales7.
Diagnóstico prenatalEn casos de familias con defecto conocido, se debe planificar la descendencia de forma preconcepcional, o lo más precozmente posible en caso contrario, con ayuda de genetistas. En estas circunstancias, el estudio es más dirigido al conocerse las mutaciones previamente. El SNF se ha relacionado con elevación de las cifras de alfafetoproteína (AFP) en líquido amniótico y sangre materna desde el segundo trimestre. Esta proteína se eleva en alteraciones estructurales, como defectos del tubo neural y de la pared abdominal. En estos casos, si la ecografía no detecta esas alteraciones, hay alta sospecha de mutaciones en NPHS1. En el caso de familias sin casos previos de SNF una elevación de AFP debe confirmarse posteriormente, ya que los portadores sanos elevan transitoriamente AFP con normalización posterior3,16. Por tanto, estos casos se deben confirmar por estudio de mutaciones.
El autor declara no tener ningún conflicto de intereses.