


Introducción. Los paragangliomas son tumores raros cuya frecuencia oscila entre 1/30.000-100.000 habitantes en la población general, siendo los del cuerpo carotídeo los más frecuentes (60%). Se estima que en el 10-50% de ellos son hereditarios, habiéndose identificado cuatro anomalías genéticas causantes. Presentamos los casos de dos hermanas con paraganglioma debido a mutación en el gen SDHD (11q23). Casos clínicos. Caso 1: mujer de 22 años con tumoración cervical bilateral, diagnosticada con resonancia magnética de paraganglioma carotídeo bilateral (tipo III de Shamblin), más extenso en el lado izquierdo. Caso 2: su hermana de 19 años presentaba paraganglioma carotídeo izquierdo tipo I de Shamblin, diagnosticado tras tomografía computarizada por absceso periamigdalino. La historia familiar muestra presencia de paragangliomas en su rama paterna, estando afectados el abuelo, el padre y cinco tíos. El estudio genético demostró una mutación del gen SDHD (11q23) en todos los afectados, siguiendo un patrón de herencia autosómico dominante con imprinting materno. En el primer caso se decidió tratamiento quirúrgico del paraganglioma izquierdo, requiriendo resección en bloque del tumor y de la carótida interna, con interposición de bypass carotidocarotídeo. En el segundo caso, se realizó exéresis simple del tumor, sin secuelas posquirúrgicas. Conclusiones. El paraganglioma familiar es una patología rara cuya causa genética se ha descrito. La identificación de los genes relacionados con esta entidad en los miembros de familias de alto riesgo, permitiría la detección y tratamiento precoz de estos tumores, reduciendo potencialmente la incidencia de morbimortalidad quirúrgica, la cual se relaciona con el tamaño y extensión del tumor. [ANGIOLOGÍA 2008; 60:127-33]
Introduction. Paragangliomas are rare tumours with an incidence ranging between 1/30,000-100,000 inhabitants in the general population, the most frequent being those affecting the carotid body (60%). Estimates suggest that 10-50% of them are hereditary, and four genetic abnormalities have been identified as causal agents. We report the cases of two sisters with paragangliomas due to a mutation in the SDHD gene (11q23). Case reports. Case 1: a 22-year-old female with bilateral cervical tumours, which were diagnosed by means of magnetic resonance imaging as bilateral carotid paragangliomas (Shamblin type III); the tumour was larger on the left side. Case 2: the first patient's 19-year-old sister presented with carotid paraganglioma (Shamblin type I) on the left side; computerised tomography scanning led to a diagnosis of a peritonsillar abscess. There was a family history of paragangliomas in the paternal branch, with the grandfather, father and five uncles all being affected by such tumours. The genetic study proved the existence of a mutation in the SDHD gene (11q23) in all those affected by the condition, which followed an autosomal dominant pattern of inheritance with maternal imprinting. In the first case, surgery was chosen to treat the left paraganglioma, involving resection of the whole tumour and the internal carotid, with placement of a carotid-carotid bypass. In the second case, simple exeresis of the tumour was performed, with no post-surgical sequelae. Conclusions. Familiar paraganglioma is a rare pathology with a genetic causation that has been described. Identifying the genes linked to this condition in members of high-risk families would allow for early detection and treatment of these tumours. This would result in a reduction in the incidence of surgical morbidity and mortality, which is related to the size and extent of the tumour. [ANGIOLOGÍA 2008; 60: 127-33]
El cuerpo carotídeo es un órgano pequeño y altamente vascularizado localizado en el tejido periadventicial del aspecto medial de la bifurcación carotídea, relacionado con el sistema paraganglionar [1].
Fisiológicamente funciona como quimiorreceptor sensitivo a los cambios en la tensión de oxígeno arterial, contenido de CO2 y concentración de ión hidrógeno y está implicado en el control de la presión sanguínea, la frecuencia cardíaca y la respiración [2].
Los paragangliomas son tumores neuroendocrinos derivados de los paraganglios extraadrenales del sistema nervioso parasimpático [3]. Son raros, con una incidencia de 1:30.000-1:100.000 en la población general [4,5]. Se estima que representan el 0,03% de todos los tumores del cuerpo y el 0,6% de los tumores de cabeza y cuello [6,7]. El más frecuente es el del cuerpo carotídeo (60-78%) [8,9]. Se presentan en la edad media de la vida como lesiones ocupantes de espacio asintomáticas, de lento crecimiento. Aunque todos los paragangliomas contienen granulos citoplasmáticos neurosecretores, solamente una minoría de ellos (1-3%) demuestra evidencia clínica de hiperfuncionalidad [3,8].
La gran mayoría son benignos. Se ha observado una conducta maligna en aproximadamente el 4-16% de ellos [3,8]. El único criterio de malignidad es la presencia de metástasis en los ganglios linfáticos cervicales o en órganos a distancia (pulmón, hígado, hueso, piel) [3,10].
Aproximadamente el 10-50% de ellos son hereditarios, con herencia autosómica dominante e imprinting materno [3,8,11,12]. Se han descrito, al menos, cuatro síndromes de paragangliomas hereditarios: PGL1, el cual resulta de una mutación en el gen SDHD (succinato deshidrogenasa subunidad D) (11q23); PGL2, en 11q13; PGL3, resultado de la mutación en el gen SDHC (1q21); y PGL4, resultado de una mutación en el gen SDHB (1p21). La mayoría de los paragangliomas hereditarios están causados por mutaciones en el gen SDHD[4].
Describimos el caso de dos hermanas con paragangliomas carotídeos familiares asociados a la mutación del gen SDHD (11q23).
Casos clínicosCaso 1. Mujer de 22 años, con antecedentes de asma, mononucleosis infecciosa hace 1,5 años, anemia en estudio y amigdalectomía, diagnosticada en el 2002 de paraganglioma carotídeo bilateral: derecho de 2,7 × 2,3 × 2cm e izquierdo de 4 × 3 × 3cm. La paciente presentaba una masa cervical lateral bilateral, mayor en lado izquierdo, con movilidad lateral y fija en el eje craneocaudal, pulsátil y no dolorosa. No refería síntomas sugerentes de hiperfuncionalidad, con estudio de catecolaminas en orina dentro de la normalidad.
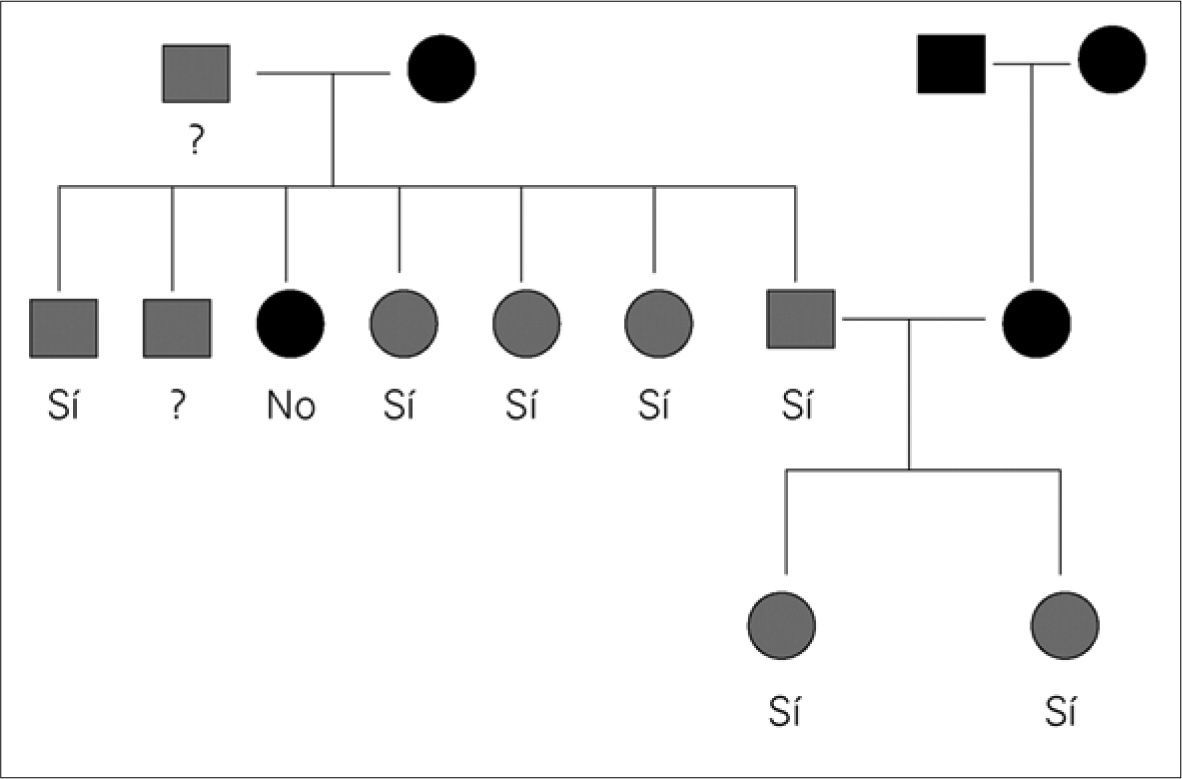
La historia familiar reveló la presencia de paragangliomas carotídeos en el abuelo paterno, padre, hermana de 19 años y en cinco de los seis tíos paternos. Se realizó el estudio genético a las dos hermanas, padre y cinco tíos (Fig. 1), encontrándose la mutación germinal R22X en el gen SDHD (11q23) en todos los afectados.

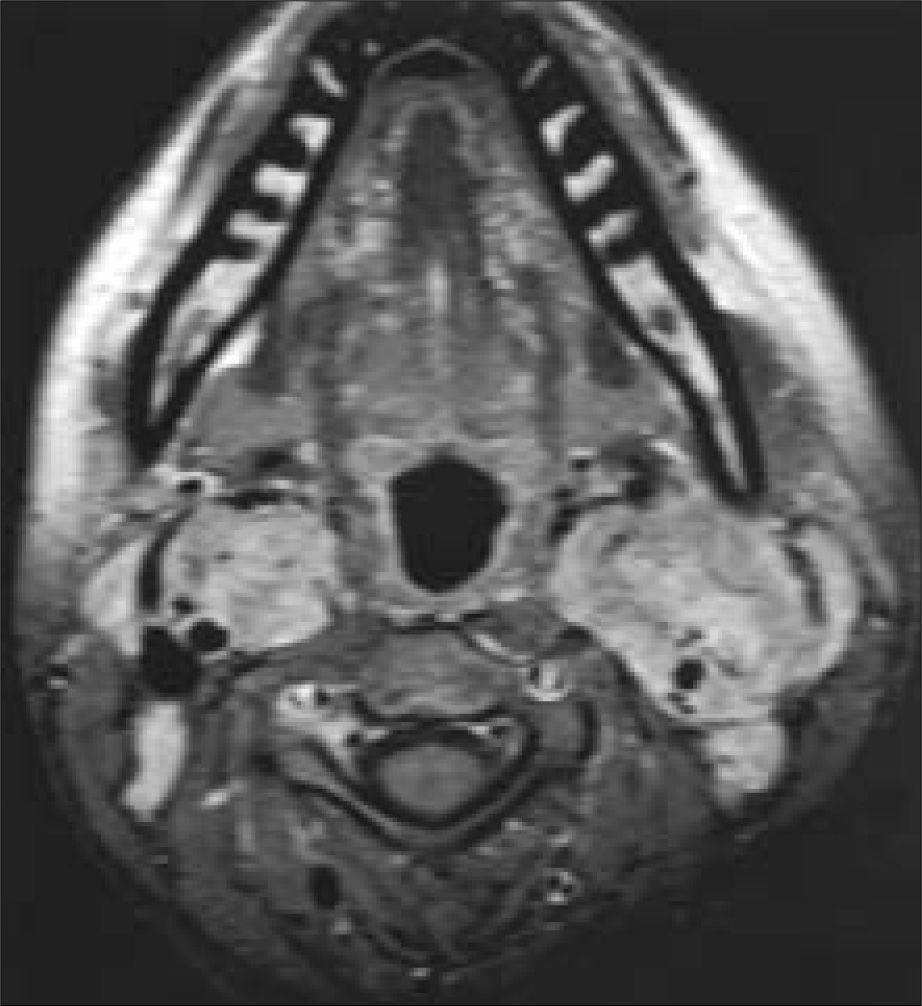
La paciente rechazó el tratamiento quirúrgico en ese momento, dado que uno de sus tíos había sufrido un accidente cerebrovascular agudo al ser intervenido en otro hospital. Se realizó seguimiento con resonancia magnética (RM), observándose un crecimiento progresivo del paraganglioma izquierdo de un 10% desde 2002 a octubre de 2004, englobando cuerpo carotídeo y rodeando carótida interna y externa (tipo III de Shamblin). El derecho se mantenía más estabilizado, englobando carótida externa casi totalmente en el borde anterior, con carótida interna desplazada posteriormente y rodeada de forma semiconcéntrica –tipo II de Shamblin– (Fig. 2).
. Paraganglioma carotídeo derecho englobando carótida externa casi totalmente en el borde anterior, con carótida interna desplazada posteriormente y rodeada de forma semiconcéntrica (tipo II de Shamblin).")
Paraganglioma carotídeo izquierdo englobando cuerpo carotídeo y rodeando carótida interna y externa (tipo III de Shamblin). Paraganglioma carotídeo derecho englobando carótida externa casi totalmente en el borde anterior, con carótida interna desplazada posteriormente y rodeada de forma semiconcéntrica (tipo II de Shamblin).
Dado el crecimiento progresivo, la paciente aceptó el tratamiento quirúrgico (primero del lado izquierdo). Se realizó arteriografía: paraganglioma cervical derecho que se rellena de contraste a partir de ramas de la arteria carótida externa, sin que exista una clara rama nutricia. Abundante colateralidad alrededor del paraganglioma. Paraganglioma cervical izquierdo mayor que el contralateral, rellenado con contraste a expensas de ramas de la arteria carótida externa, existiendo múltiples ramas nutricias de dicho paraganglioma.
En mayo de 2005, la paciente fue intervenida, objetivándose intraoperatoriamente una masa de consistencia dura de aproximadamente 5cm de diámetro que englobaba carótida común, bifurcación carotídea y carótida interna y externa, sin hallar plano de disección entre el tumor y los vasos; por ello, se realizó resección en bloque de la tumoración y de ambas carótidas más bypass con vena safena interna desde carótida común a carótida interna terminoterminal y ligadura de carótida externa, utilizando shunt recto durante la realización del bypass. Se enviaron cuatro ganglios linfáticos para estudio anatomopatológico, siendo éste negativo para infiltración tumoral.
Tras la cirugía, la paciente no presentó focalidad neurológica, aunque sí mostraba parálisis en posición paramediana de la cuerda vocal izquierda.
A las 48 horas de la cirugía, comenzó con cefalea frontal intensa, de predominio nocturno, no pulsátil y náuseas, que cedió con analgesia, sin otra focalidad neurológica. Se realizó tomografía computarizada (TC) de cráneo sin hallarse lesiones significativas. En Dúplex de bypass se objetivó su oclusión.
En el seguimiento a 16 meses, se observó una recuperación completa de la voz, con morfología y movilidad normal de las cuerdas vocales.
En las RM de control, no se visualizaron restos tumorales o recidiva en el espacio carotídeo izquierdo. Se observó un aumento del tamaño del paraganglioma derecho, con un tamaño actual de 4 × 3 × 3cm, que está pendiente de tratamiento.
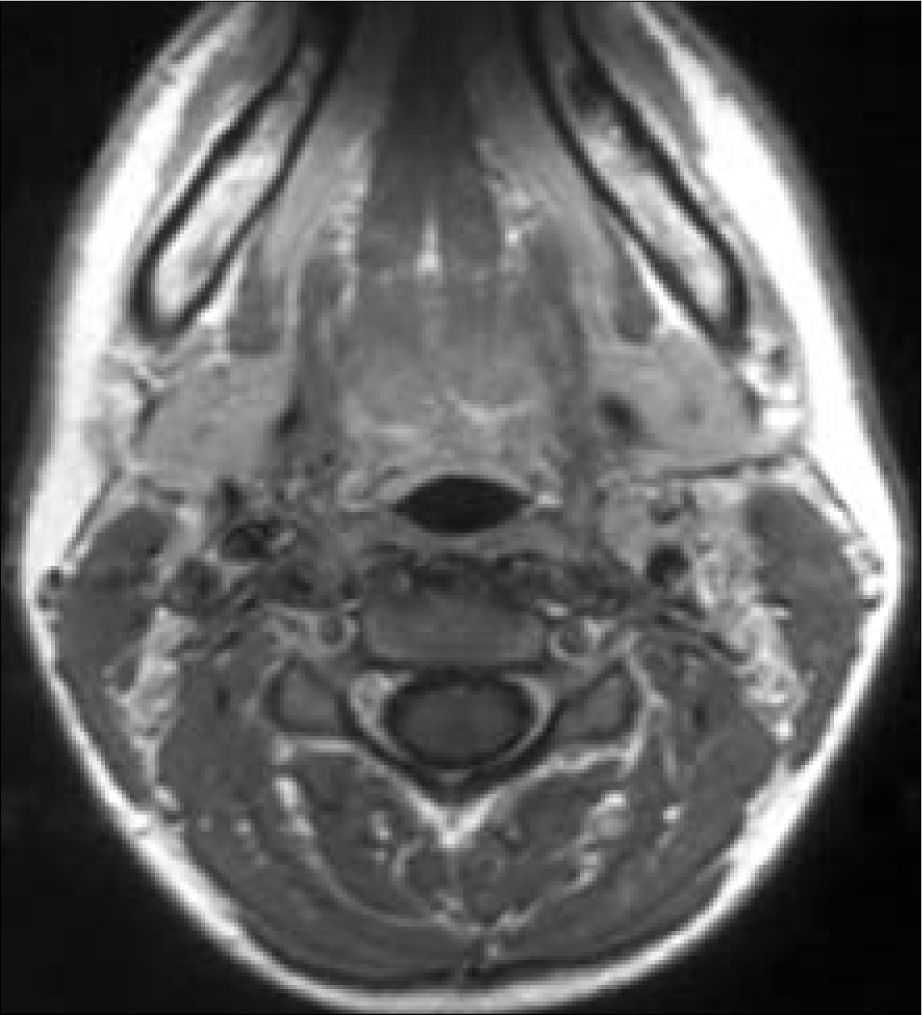
Caso 2. Mujer de 19 años, con antecedentes de adenoidectomía, absceso periamigdalino, otitis serosa y asma leve, hermana del caso anterior y portadora de la misma mutación. La paciente fue remitida a nuestras consultas por hallazgo casual de paraganglioma cervical izquierdo de unos 2,1cm de diámetro máximo en TC cervical por absceso periamigdalino en marzo de 2004. Se realizó dúplex de troncos supraaórticos (TSA), observándose un tumor glómico izquierdo que aparentemente respetaba carótida interna, de 23 × 7mm, vascularizado desde carótida externa, sin alteraciones en el lado derecho. En RM cervical se objetivó paraganglioma del cuerpo carotídeo izquierdo, de 17 × 10 × 8mm, que se localiza justo en la bifurcación carotídea, desplazando anteriormente carótida externa y posterolateralmente a carótida interna –tipo I de Shamblin– (Fig. 3). Estudio de catecolaminas en orina dentro de la normalidad.
.")
En la arteriografía preoperatoria se observó un tumor que ensanchaba la bifurcación carotídea, sin objetivarse aporte principal por ninguna rama susceptible de embolización.
La paciente fue intervenida, realizándose extirpación del paraganglioma carotídeo, sin que existieran complicaciones intraoperatorias. Tras la cirugía, la paciente permaneció neurológicamente asintomática y sin afectación de pares craneales en la exploración.
En el seguimiento a 6 meses, la paciente se encuentra asintomática.
DiscusiónLos paragangliomas del cuerpo carotídeo son los más frecuentes de los paragangliomas de cabeza y cuello (60-78%) [8,9]. Se estima que la proporción de paragangliomas carotídeos familiares oscila entre el 10 y el 50% de los casos [11]. Éstos son más frecuentemente múltiples (26-80% de los familiares [2,7,10,12] frente al 3-20% de los esporádicos [2,4,12]) y bilaterales (31,8% en los familiares frente al 4,4% en esporádicos [8]). La edad media al diagnóstico también parece ser menor en los familiares (30-35 años) que en los esporádicos (45 años) [2,13].
Se presentan como masas no dolorosas cervicales, de lento crecimiento –la media en que doblan su tamaño oscila entre 4,2 y 10 años [7,14]–, pulsátil, móvil en sentido lateral, pero no en el eje craneocaudal (signo de Fontaine). Los síntomas pueden relacionarse con la afectación de pares craneales adyacentes -IX, X, XI, XII- (0-20% de los casos) [5,9] o con la liberación excesiva de catecolaminas por parte del tumor (1-3%) [3,8].
La mayoría son benignos. La tasa de malignidad oscila en 6-30% [8] y es más común en los esporádicos [3,8]. Los criterios histológicos como pleomorfismo nuclear, atipia y actividad mitótica no son signos definitivos de malignidad. El único criterio es la presencia de metástasis en los ganglios linfáticos cervicales o en sitios a distancia (pulmón, hueso, mama, hígado) [5,8,10]. Una conducta agresiva de un paraganglioma no implica malignidad [1].
Actualmente, se han identificado cuatro loci responsables: tres de ellos codifican subunidades del complejo II mitocondrial, SDHB (PGL4) [15], SDHC (PGL3) [16] y SDHD (PGL1) [17]. El gen del PGL2 se encuentra en el cromosoma 11q13 [18] y todavía no se ha identificado [11,19]. SDHD y SDHC son subunidades de la enzima succinato dehidrogenasa, las cuales anclan a otras dos subunidades, SDHA y SDHB, en el interior de la membrana mitocondrial [4]. La succinato dehidrogenasa no sólo participa en el ciclo de Krebs, sino que también pertenece al complejo II de la cadena respiratoria mitocondrial, la cual controla el transporte aeróbico de electrones y la regulación de la concentración de oxígeno. Se ha postulado que la mutación en SDHD conduce a una estimulación hipóxica crónica, determinando una proliferación celular del tejido paraganglionar que aumenta la trascripción de VEGF (factor de crecimiento endotelial vascular) y de enzimas glicolíticas. Esto promovería la acumulación de succinato, produciendo la estabilización del HIF 1α (factor inducible por la hipoxia subunidad 1α) y la trascripción de genes conocidos implicados en la tumorogénesis, disminuyendo la apoptosis y aumentando la producción de radicales de oxígeno que activarían una vía pseudohipóxica [4,19].
En nuestros pacientes se realizó un estudio molecular del gen SDHD (11q23), a partir de una muestra de sangre periférica, mediante secuenciación automática, utilizando cebadores específicos de ADN que flanquean al exón 2, donde se localiza la mutación responsable de la enfermedad, R22X. El estudio mostró un patrón molecular alterado en los pacientes, lo que indicaba que habían heredado el cromosoma portador de la alteración. Igualmente se realizó el estudio molecular en el tumor, confirmando la mutación detectada en sangre periférica, y sugiriendo la existencia de pérdida de heterocigosidad, resultado que estaría de acuerdo con el papel que tiene asignado de gen supresor de tumores.
El gen SDHD está sujeto a una herencia autosómica dominante con imprinting materno, es decir, si el transmisor es el padre, los hijos que sean portadores de la alteración desarrollarán la enfermedad con una penetrancia del 50 y del 86% a los 31 y 50 años, respectivamente, con independencia de si son hombres o mujeres. Si el transmisor es la madre, podrá transmitir la alteración con una probabilidad del 50% en cada gestación; pero ninguno de sus hijos desarrollará la enfermedad.
En todos los pacientes se debe realizar una historia clínica personal y familiar, sospechando la presencia de mutaciones germinales en aquellos casos de edad precoz, tumores bilaterales, multicéntricos e historia familiar de paragangliomas. En estos pacientes se debe realizar un estudio genético para descartar los síndromes más frecuentes asociados con paragangliomas: gen VHL (von Hippel-Lindau), gen RET (MEN II), SDHB o SDHD (PGL). Se necesita un consentimiento informado para realizar este estudio debido las implicaciones éticas que conlleva: discriminación genética por parte de las compañías de seguros, impacto psicológico en el paciente, implicación para los familiares y la posibilidad de que el paciente no quiera revelar dicha información genética a otros familiares. En cualquier caso, una vez la mutación se ha identificado, no sólo se deberían realizar cribado de posibles tumores asociados de manera regular, sino que, además, se le debería ofrecer un estudio genético a todos los familiares de primer orden para determinar la presencia o ausencia de una mutación familiar específica [19].
En el seguimiento de los pacientes asintomáticos portadores de una mutación, actualmente se ha propuesto realizar, de manera anual, historia y exploración física, medición de tensión arterial y bioquímica, así como determinación de catecolaminas y metanefrinas fraccionadas en orina de 24 horas y/o metanefrinas fraccionadas en plasma. TC o RM de cuello, tórax, abdomen y pelvis cada 2 años, y gammagrafría con [123I]-metaidobencilguanidina cada 2-3 años. El momento en que se debe comenzar con el estudio es controvertido, aunque se suele aceptar que sea 10 años antes que el caso más precoz diagnosticado en la familia. Esto permitiría la detección de los tumores en un estadio temprano, lo cual se ha visto que reduce la morbimortalidad del posterior tratamiento [19].
En el caso de paragangliomas carotídeos bilaterales, algunos autores sugieren que primero debe resecarse el tumor más pequeño para así limitar el riesgo quirúrgico vascular y neurológico, permitiendo la cirugía del tumor contralateral. De esta manera, si tras la primera cirugía el nervio vago y el nervio hipogloso son funcionales, la cirugía contralateral podría llevarse a cabo. Por el contrario, en caso de lesión de alguno de estos nervios, no debe realizarse resección del tumor contralateral, dado que el déficit neurológico bilateral de estos nervios podría tener consecuencias clínicas graves. En estos casos, podría considerarse el tratamiento con radioterapia para el tumor contralateral [5]. Se han descrito tasas de estabilización de los tumores superiores al 96% de los casos [3,7,10]. Sin embargo, hay que tener en cuenta que el tumor persistente tras la radioterapia puede sufrir una degeneración maligna (0,28%), sobre todo en gente joven [7,10,14]. También se han descrito radionecrosis del hueso temporal (1,7%) y necrosis cerebral (0,84%) [10]. En nuestro primer caso se realizó resección en primer lugar del tumor de mayor tamaño y, aunque la paciente se ha recuperado de la lesión del nervio laríngeo recurrente, el hecho de haber sufrido una oclusión de la carótida interna en el postoperatorio y de que el tumor contralateral sea un tipo II de Shamblin, puede hacer reconsiderar si el tratamiento quirúrgico es la mejor opción de tratamiento y si, en este caso, la radioterapia puede ofrecer ventajas frente a la cirugía. Actualmente, la paciente ha rechazado el tratamiento quirúrgico por el momento, realizándose seguimiento con angio-RM.
Los paragangliomas hereditarios son tumores raros cuya genética se ha descrito, haciendo necesaria la realización de tests genéticos moleculares en aquellos pacientes con sospecha de paragangliomas asociados a mutaciones germinales y en los familiares de alto riesgo en caso de confirmación. La identificación de estos pacientes permitiría la detección y tratamiento precoz de estos tumores, disminuyendo potencialmente la morbimortalidad quirúrgica asociada a ellos, la cual parece estar relacionada con el tamaño y la extensión del tumor.
