


Los paragangliomas carotídeos (PGC) son tumores infrecuentes que muy raramente son causa de muerte. El conocimiento de las bases genéticas de los tumores de origen hereditario, más de un tercio del total, ha permitido identificar a la población en riesgo y establecer en ella medidas para un diagnóstico precoz de los tumores. El tratamiento más habitual ha sido el quirúrgico que, en no pocos casos, origina una importante morbilidad. Un amplio contingente de los PGC no experimenta crecimiento alguno, lo que justifica una conducta expectante. Aquellos tumores con crecimiento significativo deben ser tratados. La radioterapia ofrece unas cifras similares de control a las de la cirugía y no presenta secuelas neurológicas ni vasculares. No obstante, la cirugía está indicada en PGC Shamblin I/II de pacientes jóvenes, en los raros tumores malignos o hiperfuncionantes y en el fracaso de la radioterapia. Los tumores múltiples familiares deben tratarse conservadoramente.
Carotid body paragangliomas (CBP) are uncommon tumours that very rarely cause death. The genetic basis of tumours of hereditary origin (more than a third of the total) has been determined in the last few years, which has helped to identify the population at risk and to implement screening methods for an early diagnosis of tumours. The most common treatment of CBP has been surgery, which frequently causes significant morbidity. A significant number of paragangliomas do not experience any growth, which justifies a wait-and-see approach using annual image studies. Those tumours with significant growth must be treated. Radiotherapy has similar outcomes to surgery and has no neurological or vascular sequelae. However, surgery is indicated in Shamblin I/II carotid body tumours in young patients, in the rare malignant or hyper-functioning tumours, and in the failure of radiotherapy. Multiple tumours of familial origin should be treated conservatively.
Los paragangliomas (PG) se originan en el tejido paraganglionar normal del cuello que procede de las células ectodérmicas de la cresta neural. Los PG de cabeza y cuello más frecuentes son los carotídeos (60-80%), seguidos por los yugulotimpánicos (18-36%) y los vagales (3-5%)1. Los PG son tumores poco frecuentes que representan menos del 0,5% de los tumores de cabeza y cuello, aproximadamente 1-3 casos por millón de habitantes al año2. En Holanda, país donde hay una tasa relativamente alta de PG de origen genético, los paragangliomas carotídeos (PGC) tienen una incidencia anual de 0,06 casos por 100.000 habitantes3. Los PG se presentan por igual en todas las razas y áreas geográficas, aunque se ha señalado que son más frecuentes en las poblaciones que viven en altitudes elevadas, particularmente los PGC, así como en áreas con alta tasa de mutaciones fundacionales de los genes SDH, tal como ocurre en Holanda.
Los PGC se originan en el glomus caroticum, situado subadventicialmente en la bifurcación carotídea, como una masa de crecimiento muy lento. En los tumores muy grandes puede ocurrir una parálisis del vago y, menos frecuentemente, de otros pares craneales bajos. La clasificación más utilizada es la descrita por Shamblin et al.4, según la cual los PGC Shamblin de clase I muestran un simple desplazamiento de las carótidas sin apenas adherencia a la pared arterial, en los Shamblin de clase II las carótidas están parcialmente rodeadas y en los Shamblin de clase III están rodeadas en más de 270° de la circunferencia. Suárez et al.5, en una revisión de 734 casos donde constaba la clasificación, documentaron que los tumores de clase I eran el 26,6%, los de clase II el 43,9% y los de clase III el 29,6%.
Los PGC deben diferenciarse de los paragangliomas vagales (PGV), que se originan a partir del tejido glómico que existe alrededor del ganglio nudoso o plexiforme del nervio vago, justo por debajo de la base del cráneo y, aunque estos tumores suelen localizarse al completo en el compartimento retroestíleo del espacio parafaríngeo cervical, algunos pueden extenderse a la fosa posterior y al hueso temporal. A diferencia de los PGC, en los que la parálisis preoperatoria de pares bajos es poco frecuente, en los PGV casi la mitad de los casos muestran afectación de alguno de los pares craneales bajos, particularmente el vago. Otra característica diferencial es que la incidencia de malignidad de los PGV se estima en alrededor del 10-19% de los casos6,7, mientras que en los PGC está en torno al 4%5. La estructura microscópica de los PG hace imposible diferenciar los tumores benignos de los malignos, cuya naturaleza solo se puede dilucidar por la existencia de metástasis ganglionares cervicales o a distancia.
El objetivo de esta revisión es evaluar la literatura a la luz de los recientes conocimientos sobre la biología del tumor y sus bases genéticas, de forma que se pueda determinar la modalidad de tratamiento —observación, cirugía o radioterapia— más efectiva para el control del tumor y la más segura para la calidad de vida del paciente.
Diagnóstico de imagen y genéticoEn los PG, la TC con contraste muestra una masa que lo capta de forma notable, realzándolo y permitiéndonos, además, apreciar la extensión y límites de la tumoración, el grado de afectación de la base del cráneo, así como sus relaciones con las estructuras vecinas. La RM con gadolinio aporta la misma información que la TC. Es característico de estos tumores el aspecto en «sal y pimienta» por la heterogeneidad de la captación del contraste. La RM ha sustituido a la arteriografía como método de confirmación del diagnóstico, pues proporciona información sobre el tamaño, vascularización, relación con otras estructuras anatómicas y la posible presencia de tumores múltiples en el territorio cráneo-cervical. De especial interés es la información que proporciona sobre la invasión intracraneal.
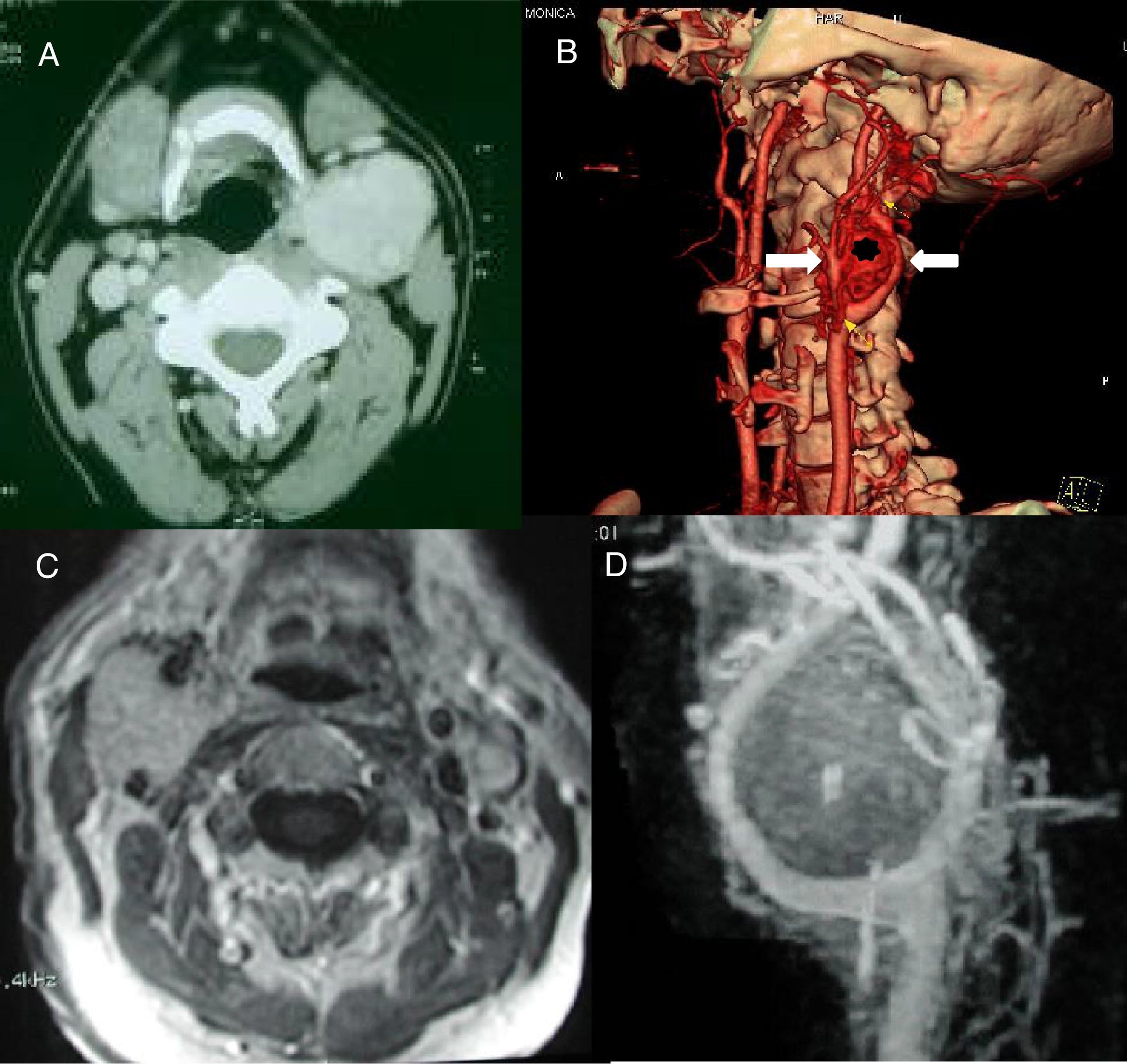
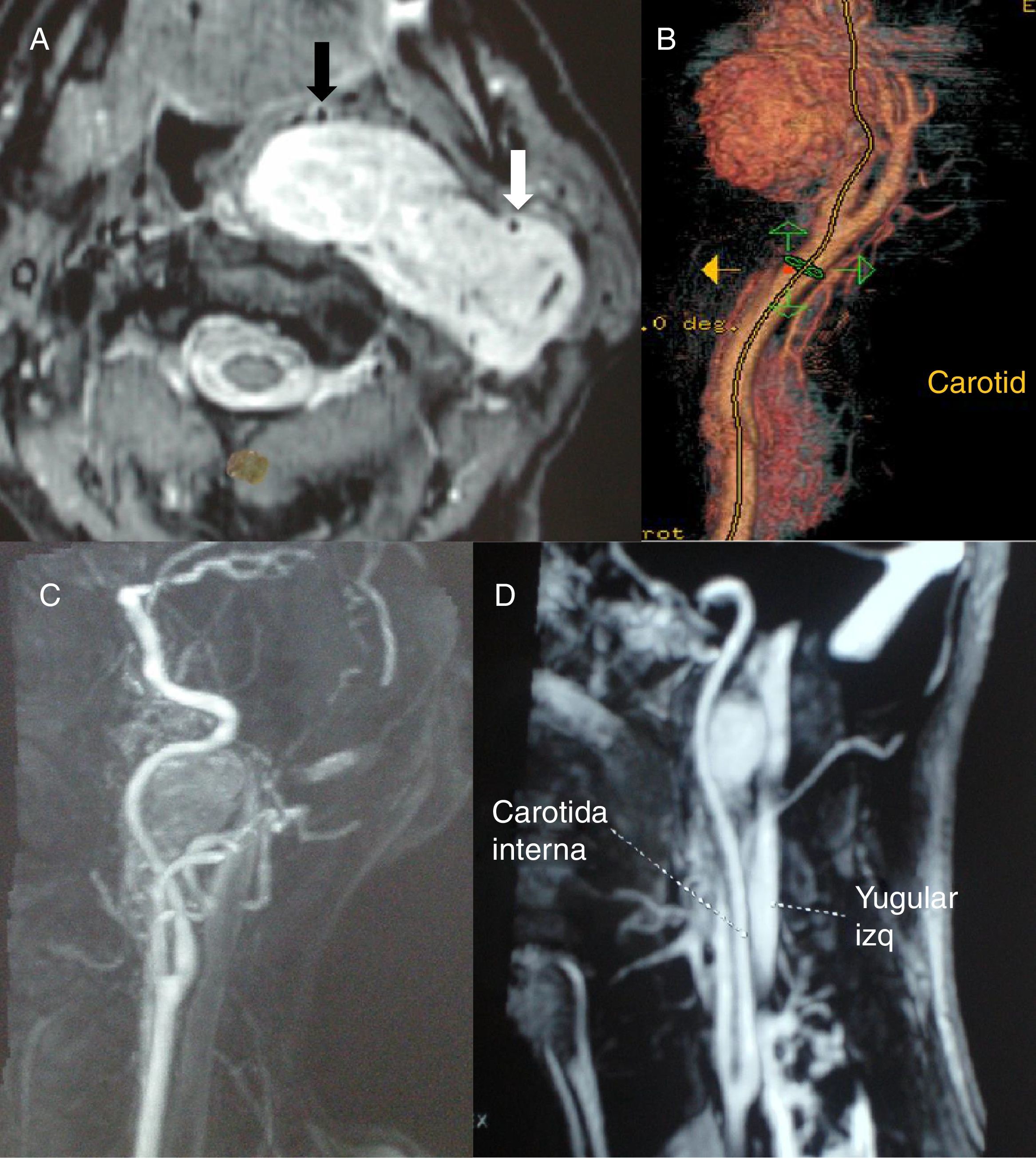
Tanto en la TC como en la RM, en los PGC la carótida externa está desplazada anteromedialmente, mientras que la carótida interna lo hace posterolateralmente, con una notable separación entre ambas (fig. 1). Por el contrario, los PGV desplazan ambas carótidas hacia delante, a la vez que comprimen y desplazan hacia atrás la vena yugular interna (fig. 2). Además, los estudios de imagen pueden anticipar con frecuencia la clasificación de Shamblin de los PGC.
 TAC con contraste. Las carótidas interna y externa están rodeadas por el tumor (posible Shambin III). B) TC tridimensional. PG carotídeo (asterisco) entre la carótida interna (flecha dcha.) y externa (flecha izda.). C) RMN con PG carotídeo izdo. interpuesto entre las carótidas interna y externa. D) Angio-RMN que muestra la ocupación del espacio entre la bifurcación carotídea por un PG.")
A) TAC con contraste. Las carótidas interna y externa están rodeadas por el tumor (posible Shambin III). B) TC tridimensional. PG carotídeo (asterisco) entre la carótida interna (flecha dcha.) y externa (flecha izda.). C) RMN con PG carotídeo izdo. interpuesto entre las carótidas interna y externa. D) Angio-RMN que muestra la ocupación del espacio entre la bifurcación carotídea por un PG.
 RMN axial en la que se aprecia un PG vagal que desplaza hacia delante las carótidas interna (flecha blanca) y externa (flecha negra). B) TC tridimensional. Las arterias carótidas aparecen desplazadas hacia delante. C) Angio-TAC que muestra el desplazamiento anterior de la carótida interna y la normalidad de la bifurcación carotídea. D.) Angio-TAC con pequeño PG vagal interpuesto entre la carótida interna y la vena yugular interna.")
A) RMN axial en la que se aprecia un PG vagal que desplaza hacia delante las carótidas interna (flecha blanca) y externa (flecha negra). B) TC tridimensional. Las arterias carótidas aparecen desplazadas hacia delante. C) Angio-TAC que muestra el desplazamiento anterior de la carótida interna y la normalidad de la bifurcación carotídea. D.) Angio-TAC con pequeño PG vagal interpuesto entre la carótida interna y la vena yugular interna.
En el estudio de los PG familiares se han utilizado también como método de «screening» compuestos radiactivos que se ligan a ciertos receptores en personas portadoras de mutación de algún gen de susceptibilidad, con la ventaja de detectar posibles lesiones sincrónicas en otros territorios. Los PG tienen una alta densidad de receptores de somatostatina de tipo 2 en la superficie de la célula, por lo que la administración de análogos de la somatostatina, como el octreótido, unidos a un radioisótopo (111indio-DTPA), producen una imagen escintigráfica de los tumores que expresan receptores de somatostatina de tipo 2 con una sensibilidad mayor del 90%. Con la misma finalidad se ha utilizado con éxito la escintigrafía con 123IMIBG, específica de los PG pero de menor sensibilidad8 y, sobre todo, la tomografía de emisión de positrones 18F-DOPA PET, que en un trabajo comparativo con los métodos de exploración anteriormente citados fue el más eficaz en la localización de estos tumores9.
Los PG pueden ser esporádicos o familiares. Las formas familiares suelen presentarse aisladas o integradas en otras entidades sindrómicas, como la neoplasia endocrina múltiple de tipo 2 (MEN2A y 2B), el síndrome de von Hippel-Lindau, la neurofibromatosis de tipo 1 y el síndrome de Carney-Stratakis, pero los originados en estos síndromes constituyen entre todos poco más del 1% de los PG. En general, se acepta que en más del 35% de los pacientes con PG existe una predisposición hereditaria, apareciendo los tumores de forma aislada. Estudios genéticos han permitido identificar los genes causantes de la enfermedad, que se localizan fundamentalmente en 5 locus: SDHD (11q23), SDHC (1q21), SDHB (1p36.1p35), SDHAF2 (11q13.1) y SDHA (5p15). Los genes SDHA, SDHB, SDHC y SDHD codifican subunidades del mismo nombre en el complejo heterotetramérico mitocondrial II, también conocido como enzima succinato deshidrogenasa (SDH), que desempeña un papel crucial en el ciclo de Krebs y en la fosforilación oxidativa, mientras que el SDHAF2 juega un importante papel en la flavinación de SDHA8,10,11. Las mutaciones germinales en los genes SDH son responsables de más del 80% de los casos familiares (51,4% mutaciones germinales en SDHD, 34,4% en SDHB y 14,2% en SDHC en una serie de 183 casos con mutación)12. Los pacientes con mutaciones del gen SDHD presentan el riesgo más alto de desarrollar PG a lo largo de la vida, con una altísima penetrancia (cerca del 90%)13. Así, se ha demostrado una prevalencia de PG asintomáticos del 59% en portadores de esta mutación mediante RMN14. Además, los PG múltiples son un hecho habitual de este síndrome: ocurren en el 60-80% de los pacientes, ya sea de forma sincrónica o metacrónica. Igualmente, las mutaciones en SDHD pueden predisponer a PG extracervicales y feocromocitomas (FCC) (15-20% de los casos)15.
Aunque muy escasos, los pacientes con PG por mutación en SDHAF2 comparten con los anteriores las características de multicentricidad y alta penetrancia10.
Por el contrario, los pacientes con mutaciones en SDHB desarrollan frecuentemente PG extraadrenales (50-80%) y FCC (20-30%) y, menos frecuentemente, PG de cabeza y cuello (30-40%), que son generalmente únicos, con una penetrancia intermedia de esta enfermedad11–13. Es de reseñar que en series muy amplias y revisiones de la literatura entre el 20 y el 41% de las mutaciones en SDHB se asocian a un PG o FCC maligno12,16, por lo que tienen peor pronóstico. Los tumores por mutación del gen SDHC son, en su mayor parte, únicos y de localización cervical, con una baja penetrancia12.
La mutación del gen SDHA causa generalmente una enfermedad familiar neurológica llamada síndrome de Leigh y solo de forma excepcional PG de cabeza y cuello o FCC (2,2%)17. Otros genes de susceptibilidad de PG familiares recientemente descubiertos son TMEM127 y MAX, pero su incidencia es muy baja8,11. Cerca de un tercio de los PG sin historia familiar, por tanto, aparentemente esporádicos, presentan también alteraciones germinales en SDH (formas familiares ocultas). En los casos esporádicos restantes se desconoce la alteración causante.
Los tumores asociados a mutaciones de los genes SDHB y SDHC se transmiten de forma autosómica dominante, indistintamente sea del padre o de la madre, pero cuando los causantes son genes SDHD y SDHAF2 la herencia es de tipo autosómico dominante, modificada por impronta genética, de tal forma que no hay expresión del gen cuando se hereda de la madre, mientras que su transmisión por parte del padre resulta en enfermedad en los hijos12,13,18, lo que explica que haya saltos generacionales. Una característica de los PG de origen genético es su aparición a edades mucho más tempranas que los esporádicos.
Basándose en las citadas correlaciones entre genotipo-fenotipo, se pueden hacer una serie de recomendaciones para el «screening» genético, que por razones de coste deben basarse inicialmente en los genes más comúnmente alterados12.
Por otra parte, la inmunohistoquímica de SDHB es un excelente indicador de las mutaciones germinales de los genes SDH y reduce los costes del «screening» genético. En las células de los PG esporádicos el SDHB mitocondrial se tiñe intensamente, mientras que en los tumores con mutaciones en los genes del sistema SDH, la tinción de SDHB disminuye mucho o desaparece, de forma que solo estos se someten al «screening» mutacional19.
Cuando se hace en un paciente el diagnóstico de un PG de origen genético, se debe comprobar si esa mutación existe en los miembros directos de la familia, pues la posibilidad de desarrollar la enfermedad es alta en aquellos portadores de la mutación. A fin de detectar la enfermedad en estadios precoces, se deben realizar pruebas de imagen anuales en los portadores de mutaciones SDHD/SDHAF2 por vía paterna y en todos los portadores de mutaciones SDHB/SDHC. Las pruebas más recomendables son el 18F-DOPA PET, el 111indio pentetreótido con imagen SPECT, o bien la RMN8,11,20.
Actitud terapéuticaEvolución biológica de la enfermedad y modalidades terapéuticasEl paradigma de la medicina actual es conseguir, siempre que sea posible, un tratamiento individualizado que, curando la enfermedad, cause las menores alteraciones posibles a la vez que mejore la calidad de vida del paciente. Un ejemplo incuestionable de esta tendencia lo constituyen los PG de cabeza y cuello, hasta hace no muchos años considerados de tratamiento quirúrgico exclusivo.
La mayoría de los PG tienen un patrón de crecimiento indolente, por el que aquellos que crecen tienen un incremento medio del tamaño tumoral menor de 1mm por año21, duplicándose el tamaño del tumor entre 4 y 14 años22,23, lo que no quita para que una pequeña parte tenga una tasa de crecimiento relativamente alta, sobre todo los malignos, que son menos del 5% del total24. En este sentido, Langerman et al.25 observaron en un grupo de 47PG cervicales (28 carotídeos y 19 vagales), seguidos durante una media de 5 años, que 19 tumores permanecieron con un tamaño estable (42%), 17 crecieron (38%), y 9 disminuyeron de tamaño (20%). De los 17 tumores que crecieron, el crecimiento medio fue de 0,2cm/año. Por ello, la observación mediante controles anuales de RM es la primera opción; se procede al tratamiento (quirúrgico o radioterápico) en los casos que haya crecimiento del volumen tumoral >20% en un año.
Un punto de gran interés es que no parece haber diferencias significativas en la supervivencia de pacientes tratados y aquellos en los que se ha seguido una conducta de «wait and see»26. Las muertes por un desarrollo incontrolado de los PG de cabeza y cuello son raras, no llegan al 4%, aun considerando los tumores malignos7,27. Estos resultados ponen en evidencia si la evolución natural de la enfermedad se mejora realmente por la intervención quirúrgica, ya que en ciertas localizaciones de mayor riesgo de complicaciones, el objetivo del tratamiento debería ser reducir la morbilidad antes que aumentar la supervivencia. Así, en caso de aumento del número de pares afectados y crecimiento intracraneal compresivo, estaría justificada la intervención. Se puede argumentar que el control anual por RM puede incrementar el riesgo de afectación de pares craneales. No parece ser así, pues aun en el caso de los PGV, que son los que más parálisis de pares bajos muestran, solo el 7,5% de los pacientes en los que se siguió este protocolo mostraron un nuevo déficit neurológico tras un seguimiento medio de cerca de 10 años28.
Existen 2modalidades de tratamiento en los PG de cabeza y cuello con un crecimiento significativo: la cirugía y la radioterapia, con sus variantes de IMRT y radiocirugía estereotáxica hipofraccionada (CyberKnife). La radioterapia ha sido mucho más utilizada en los PG de más difícil acceso quirúrgico que en aquellos de localización cervical, pero sus resultados pueden ser extrapolables a estos últimos. Así, en un metaanálisis sobre 2.042 pacientes con PG temporales tratados con cirugía o radioterapia (externa o estereotáxica), Suárez et al.7 observaron un control a largo plazo de la enfermedad en el 78,2; 89,1 y 93,7% de los tratados con cirugía, radioterapia externa y radiocirugía, respectivamente. La tasa de complicaciones graves posquirúrgicas (neumonías por aspiración, infarto cerebral, meningitis, etc.) fue significativamente superior a la de la radioterapia externa (radionecrosis) y la de esta a la de la radiocirugía. Otro aspecto relevante es el referido a las parálisis de pares craneales, que con la cirugía se duplicaron respecto a las existentes en el preoperatorio. Por el contrario, con la radioterapia hubo una discreta mejoría del estatus neurológico.
De forma análoga, en un metaanálisis similar sobre PGC, aun cuando el número de pacientes radiados era pequeño (127 vs. 2.175 operados), Suárez et al.5 no apreciaron diferencias en las cifras de control local entre la cirugía y la radioterapia, que en ambos casos estuvo en torno al 94%. Sin embargo, la cirugía representó un aumento del 22% en el déficit de pares craneales. Más aún, se resecó, con o sin sustitución, la carótida interna o común, por lesión quirúrgica o englobamiento por el tumor, en el 13% de los casos, lo que se tradujo en un 3% de accidentes cerebrovasculares permanentes y un 1,3% de mortalidad.
Finalmente, en una serie de 156PG de varias localizaciones de cabeza y cuello tratados con radioterapia, la tasa de control local a los 10 años fue del 96%, con resolución del 14% de las parálisis de pares craneales, sin que se reseñara ninguna complicación importante29.
No obstante lo que precede, la radioterapia tampoco está exenta de efectos secundarios. Así, la prevalencia de estenosis carotídea asintomática superior al 50% del diámetro oscila entre el 2 y el 8% en la población general30. En contraste, dicha tasa asciende al 30-40% en los pacientes que han recibido radioterapia en el cuello, con efecto acumulativo a largo plazo31. Se ha estimado que la radioterapia eleva el riesgo de padecer un infarto isquémico cerebral 5,6 veces sobre la población general y 9,8 veces en el segmento de menos de 50 años32.
Otro factor que considerar es el riesgo de sarcoma radioinducido, que se estima en menos del 1% a los 10 años de la irradiación33. No obstante, tanto en uno como en otro caso no se ha publicado que la radioterapia haya producido las citadas consecuencias, posiblemente por la menor dosis utilizada o porque, al contrario que en la cirugía, la complicación se desarrolla muchos años más tarde34.
Indicaciones para la cirugíaLos PGC de clase Shamblin III se asocian estrechamente a secuelas neurológicas y complicaciones vasculares en comparación con los de estadio inferior, que son del 2,3% en los tumores de clase I/II y del 35,7% en los de clase iii35.
En una revisión sobre 1.181 casos publicados de PG carotídeos, Anand et al.36 contabilizaron un 66% de incidencia de infarto cerebral con una tasa del 46% de mortalidad en 89 casos en los que se ligó la arteria carótida interna, comparado con un 9,7% de incidencia de infarto y un 2,4% de mortalidad en los 124 casos en que se reconstruyó la arteria.
Igualmente, una revisión de 1.988PG carotídeos por Vogel et al.37 mostró que los pacientes con reconstrucción de la carótida tuvieron una mayor tasa de mortalidad (1,6 vs. 0,6%), infarto cerebral (17,7 vs. 3,5%) y hemorragia posoperatoria (43 vs. 2,4%) con relación al resto de los pacientes, además de una mayor tasa de parálisis de pares craneales. Por otro lado, se ha estimado que los pacientes que reciben una extirpación del tumor con reconstrucción carotídea tienen 6 veces más probabilidades de desarrollar un infarto cerebral que aquellos en los que se deja intacta la arteria38. Todo ello aconseja abstenerse de la cirugía en los tumores de clase III, salvo en circunstancias especiales (malignidad o hipersecreción), y limitarla al resto de los estadios cuando se observe crecimiento del tumor. En el caso de decidir un tratamiento quirúrgico, la técnica más recomendable es la resección subadventicial, espacio avascular entre las carótidas y el tumor. El comenzar la disección en dirección craneocaudal39 facilita la disección de los pares craneales y disminuye la morbilidad; a veces, en los tumores grandes, es necesaria la ligadura de la carótida externa y la vena yugular interna para facilitar la extirpación.
Los PGC bilaterales son generalmente de causa genética, de forma que en una serie de 68PG carotídeos bilaterales el 76% eran hereditarios39.
Por el contrario, cuando se presentan en personas que viven a más de 2.000 m sobre el nivel del mar la mayor parte de ellos son debidos a factores que conducen a una hipoxia crónica40. La extirpación de los tumores en ambos lados produce una denervación de los senos carotídeos que tiene como consecuencia un fallo barorreceptor agudo, que se manifiesta de forma permanente en hasta el 20% de los pacientes, y que produce hipertensión lábil severa, cefaleas, diaforesis e inestabilidad emocional, por lo que se debe evitar si no hay causa que lo justifique41.
Como se ha dicho anteriormente, las mutaciones del gen SDHD predisponen a PG múltiples en otras localizaciones de cabeza y cuello, por lo que en estos casos hay que abstenerse de la cirugía, a fin de evitar el riesgo de parálisis bilaterales de pares bajos.
Los PG malignos, cuando solo presentan metástasis ganglionares, algo más frecuentes que las metástasis a distancia, son una indicación quirúrgica absoluta a fin de evitar su difusión sistémica. La supervivencia a 5 años de los PG malignos es del 60-72%5,24,42.
Los PG secretores de catecolaminas representan solo el 3-5% de todos los PG y pueden ser silentes, por lo que es necesario estudiar las catecolaminas. Los gránulos secretores de las células principales no desaparecen con la radioterapia, y persiste la secreción de catecolaminas43. En estos casos, solo la cirugía consigue normalizar los síntomas cardiovasculares y los niveles de catecolaminas.
Por último, en aquellos raros casos en los que pese al tratamiento con radioterapia el tumor siga creciendo, es necesaria la cirugía de rescate.
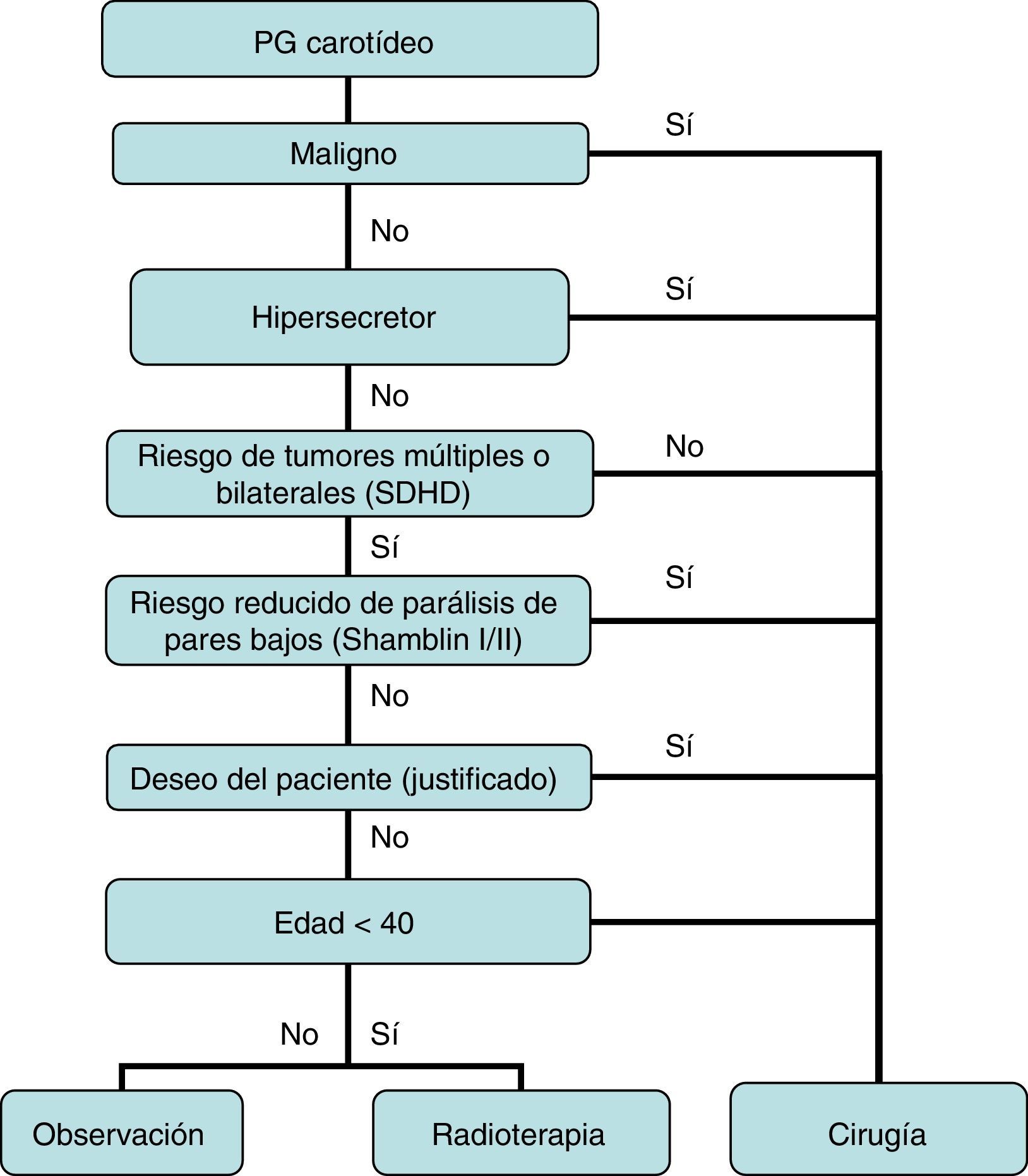
Recientemente, Lozano Sánchez44 en una documentada revisión, en la que se expone el cambio de actitud terapéutica respecto a los PGC en los últimos 10 años, ha comentado el decálogo de circunstancias que deben guiar la indicación quirúrgica y propuesto un algoritmo basado en dichas premisas, con el que en buena medida coincidimos, si bien nuestros criterios son algo más restrictivos (fig. 3).
Conclusiones
Los PGC son generalmente tumores benignos que no afectan apenas la expectativa de vida de aquellos que los padecen, lo que en principio avala observar su evolución antes de decidir un tratamiento, que se debe circunscribir a los tumores que muestran crecimiento significativo. Dado que la cirugía es causa de importante morbilidad, se debe limitar a las de clases Shamblin I/II, particularmente cuando se trata de pacientes jóvenes. En el resto es preferible utilizar la radioterapia de intensidad modulada a dosis de 45Gy o la radioterapia estereotáxica hipofraccionada (CyberKnife). Una excepción son los tumores malignos o hiperfuncionantes, cuya única indicación es la quirúrgica. Finalmente, es de suma importancia el análisis genético de los pacientes con PGC, ya que más de un tercio son de origen familiar. En caso de mutación de un gen de susceptibilidad, es preceptivo el estudio de la familia a fin de detectar a los portadores de la mutación y poder realizar un diagnóstico precoz del tumor.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
