A pro-oncogenic intestinal microbiome was observed in murine models; however, no specific microbiome in patients with hepatocellular carcinoma (HCC) has been reported. We aimed to compare the gut microbiome found in cirrhotic patients with or without HCC.
Materials and methodsFrom 407 patients with Child Pugh A/B cirrhosis prospectively followed, 25 with HCC (cases) were matched with 25 without HCC (wo-HCC) in a 1:1 ratio according to age, gender, etiology, Child Pugh and severity of portal hypertension. In addition, results were also compared with 25 healthy subjects. Fecal stool samples were sequenced for the V3–V4 region of the microbial 16S rRNA (Illumina MiSeq Platform). Plasma cytokines were quantified including interleukin-6 (IL-6) and tumor necrosis factor α (TNF-α).
ResultsWe found a differential abundance in family members of Firmicutes with a 3-fold increase of Erysipelotrichaceae and a 5-fold decrease in family Leuconostocaceae in HCC when compared to wo-HCC controls. Genus Fusobacterium was found to be 5-fold decreased in HCC vs wo-HCC. The ratio bacteriodes/prevotella was increased in HCC. Three operational taxonomic units (OTUs), genus Odoribacter and Butyricimonas were more abundant in HCC, whereas a decreased abundance in Lachnospiraceae family genus Dorea was observed in HCC patients. A Random Forest model trained with differential abundant taxa correctly classified HCC individuals. This pattern was associated with an inflammatory milieu with a putative increased activation of NOD-like receptor pathways.
ConclusionWe found a pattern of microbiome linked to inflammation that could be potentially useful as HCC biomarker after follow-up validation studies.
alpha-fetoprotein
cryptogenic cirrhosis
confidence interval
computerized tomography
hepatocellular carcinoma
Hazard ratio
interquartile range
liver transplantation
Milan criteria
Model for End-stage Liver Disease
magnetic resonance imaging
non-alcoholic fatty liver
non-alcoholic fatty liver disease
non-alcoholic steatohepatitis
percutaneous ethanol injection
radiofrequency ablation
trans-arterial chemoembolization
Increasing interest has been focused during the last years in microbiome and human diseases including cirrhosis, alcoholic liver disease, fatty liver and fibrosis progression. Progressive changes in the gut microbiome have been observed accompanying the progression of liver disease [1–4]. A reduced abundance of taxa considered benign, such as Lachnospiraceae, Ruminococcaceae and Clostridialies and a higher abundance of non-beneficial taxa such as Enterobacteriaceae and Bacteriodaceae were observed [2,5].
A renewed novel research has focused on microbiome and cancer development. However, scarce data has been written regarding microbiome and hepatocellular carcinoma (HCC) [6,7]. Previous studies showed that the severity of liver disease is linked to an inflammatory pro-oncogenic microenvironment from the intestine to the liver in murine models [8–10]. These inflammatory signals derived from changes in the intestinal microbiome have been proposed as a novel carcinogenic mechanism [9–11]. This gut-liver axis may also become a new biomarker and a potential preventive therapeutic target. Therefore, the aim of the present study was to compare the gut microbiome found in patients with cirrhosis with and without HCC to observe if differences in gut microbiome profiles were present among these patients.
2Materials and methods2.1Study design, setting and participating centersThis observational case-control study was nested on a prospective longitudinal cohort of patients with cirrhosis who were followed-up in our Liver Unit at Austral University Hospital, School of Medicine, in collaboration with HERITAS (Rosario), CONICET and the National Academy of Medicine from Argentina. This study was carried out between December 2015 and October 2016 in accordance with international recommendations for observational studies.
2.2Eligibility criteriaA consecutive non-probability sampling of adult subjects (>17 years old) with clinical or histological diagnosis of cirrhosis, functional status Child Pugh class A or B was included. Clinical or histological diagnosis of cirrhosis was done according to international consensus guidelines [13]. Exclusion criteria consisted of any of the following: (1) Patients under any immunosuppressive treatment; (2) prior or current treatment with any pre- or probiotic; (3) active alcoholism (cessation of alcohol intake at least 3 months prior to study entry); (4) past or present history of neoplasms; (5) active infection grade >2, according to the NCI CTCAE criteria, version 4.0 [14]; patients with chronic hepatitis B or hepatitis C were allowed to be included, (6) infection with human immunodeficient virus (HIV); (7) any antibiotic treatment should have been completed one month prior to the inclusion, excluding primary or secondary prophylaxis for bacterial infections in cirrhosis; (8) diarrhea secondary to any commensal, including Clostridium Difficile diarrhea within 6 months prior to the inclusion of the subject in the study; and (9) any malabsorption disorder, celiac disease or inflammatory bowel disease including ulcerative colitis and Crohn's disease.
Dietary habits were precluded in all patients including high fat diets and alcohol consumption prior the inclusion to this study. Moreover, we precluded the inclusion of vegetarian individuals or any food restrictions such as gluten or lactose and all patients were recommended to have a balanced homogeneous nutrition.
All cases met the above eligibility criteria plus imaging or histological diagnosis of HCC. Imaging HCC diagnosis was performed with a tri-phase dynamic study, either computerized axial tomography (CT) or magnetic resonance imaging (MRI) as recommended by international guidelines [15]. All cases were allowed to have a history of prior HCC treatment, excluding liver transplantation. They should have at least one active HCC nodule at time of fecal stool sample collection. Images were centrally reevaluated by a single observer, blinded from clinical and exposure variables.
Every case was matched with one control without HCC (wo-HCC) in a 1:1 ratio according to age, gender, etiology of liver disease, Child Pugh score and presence of clinically significant portal hypertension. Exclusion of HCC in all controls was done with either CT or MRI scans during the screening period. Selection process of cases and controls was prior to stool samples collection and was blinded from results of gut microbiome. In addition, we studied a gut microbiome dataset of 25 matching healthy controls [16].
2.3Exposure variables: definition and measurementThe following measurements were recorded at baseline in each subject enrolled: demographic data, concomitant medications including use of lactulose, rifaximin or norfloxacin, blood tests including serum alpha-fetoprotein (AFP) and complete physical examination [6].
Subjects with HCC diagnosis were staged according to Barcelona Clinic Liver Cancer staging (BCLC) [15] at study entry.
As the disturbance in the equilibrium of some selective key cytokines has been reported in cirrhotic patients and also because previous reports showed robust correlations between specific bacterial families and inflammatory cytokine levels, we measured IL-6 and TNF alpha levels in plasma samples to improve understanding of their role. Cytokine measurement including interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) was performed in serum obtained from peripheral blood samples at study entry on the same day of fecal samples collection. Commercial reagents based on enzyme linked immunosorbent assay (ELISA) were used (BD-Bioscience, San Diego, California, United States). Duplicate measurements were performed for every sample and expressed as pg/mL. Runs were also done blinded from clinical and other exposure variables.
2.4Fecal stool samples collection, DNA extraction and sequencingFecal samples were obtained noninvasively in a plastic collection kit at any time during the day. All samples were stored at −70°C. Each fecal sample was aliquoted for final processing in HERITAS. Total DNA extraction from stool samples (about 200mg) was performed using QIAmp DNA Stool Mini Kit following manufacture's instructions. The 16S rRNA V3-V4 hypervariable region was first amplified using PCR method (20 cycles) and then a second reading for sample identification (6 cycles). Amplicons were cleaned using Ampure DNA capture beads (Argencourt-Beckman Coulter, Inc.) and quantified using Quanti-iTTM PicoGreen® DNA Assay Kit (Invitrogen Molecular Probes, Inc., Eugene, OR, United States) with the standard protocol (high range curve – half area plate) and pooled in molar concentrations before sequencing on the Illumina MiSeq platform (Illumina, Inc., San Diego, CA, United States) using 2×300 cycles PE v3 chemistry. In each procedure, 3 measurements were made to avoid information bias. The operators of this measurement were blinded to clinical and other exposure variables.
2.5Sequence data pre-processing, classification and taxonomic assignmentAmplicon sequencing produced 11639500 raw paired-end (PE) reads. We followed the 16S SOP described in Microbiome Helper [17]. FastQC (v0.11.5) was used to analyze the raw data quality of PE reads. Paired-end reads were stitched together using PEAR (v0.9.10). Stitched reads were filtered by quality and length, using a quality score cut-off of Q30 (phred quality score) over 90% of bases and a minimum length of 400bp. Concatenated and filtered fastq sequences were converted to fasta format and we removed sequences that contain contained “N”. Potential chimeras were identified using the UCHIME algorithm, and then the chimeric sequences were removed. The remaining sequences were clustered to operational taxonomic units (OTUs) at 97% similarity level with open reference strategy implemented in Quantitative Insights into Microbial Ecology (QIIME; v1.91), using SortMeRNA for the reference picking against the Greengenes v13_8 97% OTU representative sequences database and SUMACLUST for de novo OTU picking.
2.6Statistical analysis and 16S rRNA bioinformatics processingA statistical two-tailed value, α type I error of 5% (p value<0.05) was considered. Clinical data was analyzed using STATA version 10.1.
Bioinformatics analysis of the data was done using a custom QIIME pipeline and the R package phyloseq (https://github.com/joey711/phyloseq). OTU table was rarefied at 12,566 sequences per sample for alpha and beta diversity and random forest analysis. To provide alpha diversity metrics, we calculated observed species, Chao1 and Shannon's diversity index. To evaluate beta diversity among cases, controls and healthy individuals UniFrac (weighted and unweighted) distances and Bray Curtis dissimilarity were used, prior removal of OTU's not present in at least 5% of samples. The UniFrac and Bray Curtis measures were represented by two dimensional principal coordinates analysis (PCoA) plots. Differences between groups were tested by a permutation multivariate analysis of variance (PERMANOVA) using distance matrices function (ADONIS) implemented in the R vegan package [18]. The evaluation of beta diversity between cases and controls were also determined using a principal fitted components for dimension reduction in regression as it was used to analyze microbiome data [16–18].
To understand the differences in the composition between groups, the R package DESeq2 was used to perform differential abundance estimates. Analysis of composition of microbiomes (ANCOM) was carried out with the ANCOM R package with 5% FDR [19].
We build a Random Forest model with the random Forest R package using OTU's relative abundance as features to predict samples from HCC or wo-HCC groups, measuring the “out of bag” (OOB) error to estimate the error model. From the list of variables, measured by the mean decrease in Gini index, we selected the three OTU's that contributed the most to the group classification. A random forest was trained with these 3 OTU's. To determine the model significance a permutation test with 1000 permutations was run.
2.7Functional assessment of the microbiome profileTo evaluate the impact of any observed differences in the microbiome composition we further performed an indirect functional assessment to infer the metagenomic composition with PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States). We removed OTU's not present in the database used for OTU picking, the OTU table normalized by predicted 16S copy numbers was used to predict KEGG orthologs (KOs) collapsed to KEGG Pathways. This PiCRUST analysis included any association with NOD-like receptor (NLR) signaling pathway [20]. NOD-like receptors are intracellular sensors with central roles in innate and adaptive immunity [20].
2.8Ethical considerationsThis study protocol was performed according to national standards for ethical, legal and regulatory requirements, international ethical standards according the 2008 Helsinki Declaration and its amendments from the Nuremberg code, as well as GCP standards (“Good Clinical Practice”). All the patients signed a written informed consent prior to the inclusion in this study. The study protocol was approved by the Ethics Committee from the Austral University Hospital School of Medicine (Ref number 15-073).Study protocol followed the STROBE guidelines [12]
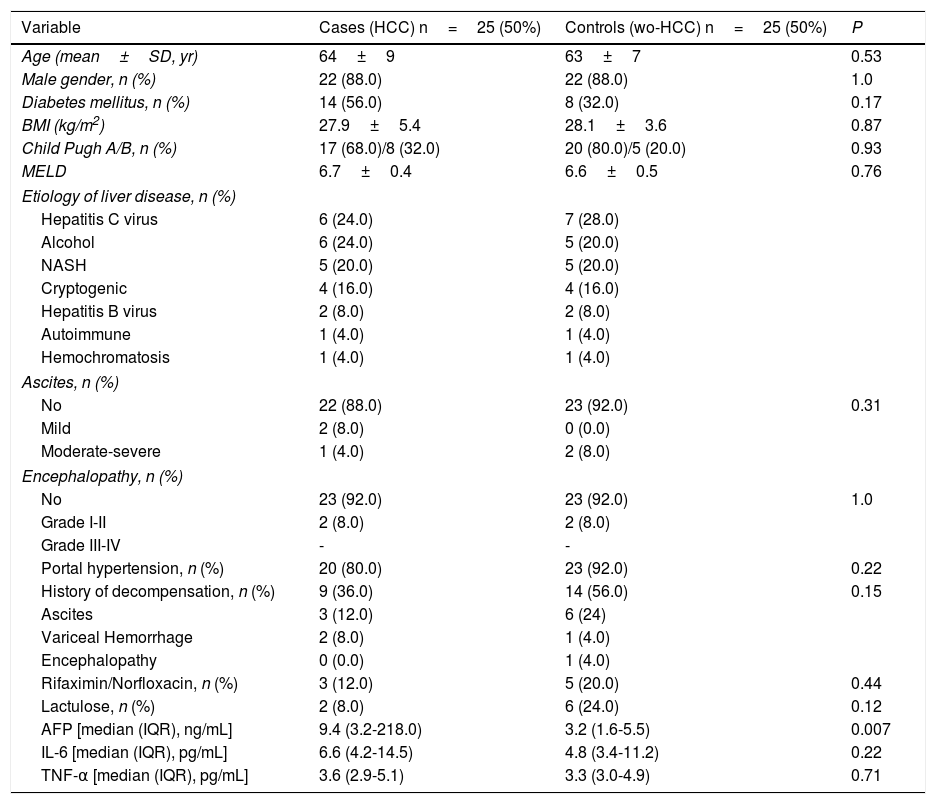
3Results3.1Participating patients characteristicsFrom a total 407 patients who were prospectively followed, 25 cases and 25 controls (wo-HCC) were included in the study. A subgroup of 25 healthy individuals was also enrolled, matched according to gender in a 1:1 ratio. All of the patients with HBV infection were under viral treatment and all the patients with HCV were viremic (none of them were treated before fecal samples were obtained). In patients with HCC, considering BCLC stages, 2 patients were in stage 0, 12 patients were in stage A, 5 were in stage B and 6 in stage C-D. Vascular invasion was observed in 3 patients and extrahepatic spread in 4. Previous HCC treatments consisted of radiofrequency ablation (RFA) in 1 patient, liver resection (LR) in 3, trans-arterial chemoembolization (TACE) in 17 and sorafenib in 3 patients. Cases and wo-HCC controls were well matched (Table 1). Although not statistically significant, higher concentrations of both inflammatory cytokines were observed in HCC patients.
Baseline clinical and laboratory variables among cases and controls.
| Variable | Cases (HCC) n=25 (50%) | Controls (wo-HCC) n=25 (50%) | P |
|---|---|---|---|
| Age (mean±SD, yr) | 64±9 | 63±7 | 0.53 |
| Male gender, n (%) | 22 (88.0) | 22 (88.0) | 1.0 |
| Diabetes mellitus, n (%) | 14 (56.0) | 8 (32.0) | 0.17 |
| BMI (kg/m2) | 27.9±5.4 | 28.1±3.6 | 0.87 |
| Child Pugh A/B, n (%) | 17 (68.0)/8 (32.0) | 20 (80.0)/5 (20.0) | 0.93 |
| MELD | 6.7±0.4 | 6.6±0.5 | 0.76 |
| Etiology of liver disease, n (%) | |||
| Hepatitis C virus | 6 (24.0) | 7 (28.0) | |
| Alcohol | 6 (24.0) | 5 (20.0) | |
| NASH | 5 (20.0) | 5 (20.0) | |
| Cryptogenic | 4 (16.0) | 4 (16.0) | |
| Hepatitis B virus | 2 (8.0) | 2 (8.0) | |
| Autoimmune | 1 (4.0) | 1 (4.0) | |
| Hemochromatosis | 1 (4.0) | 1 (4.0) | |
| Ascites, n (%) | |||
| No | 22 (88.0) | 23 (92.0) | 0.31 |
| Mild | 2 (8.0) | 0 (0.0) | |
| Moderate-severe | 1 (4.0) | 2 (8.0) | |
| Encephalopathy, n (%) | |||
| No | 23 (92.0) | 23 (92.0) | 1.0 |
| Grade I-II | 2 (8.0) | 2 (8.0) | |
| Grade III-IV | - | - | |
| Portal hypertension, n (%) | 20 (80.0) | 23 (92.0) | 0.22 |
| History of decompensation, n (%) | 9 (36.0) | 14 (56.0) | 0.15 |
| Ascites | 3 (12.0) | 6 (24) | |
| Variceal Hemorrhage | 2 (8.0) | 1 (4.0) | |
| Encephalopathy | 0 (0.0) | 1 (4.0) | |
| Rifaximin/Norfloxacin, n (%) | 3 (12.0) | 5 (20.0) | 0.44 |
| Lactulose, n (%) | 2 (8.0) | 6 (24.0) | 0.12 |
| AFP [median (IQR), ng/mL] | 9.4 (3.2-218.0) | 3.2 (1.6-5.5) | 0.007 |
| IL-6 [median (IQR), pg/mL] | 6.6 (4.2-14.5) | 4.8 (3.4-11.2) | 0.22 |
| TNF-α [median (IQR), pg/mL] | 3.6 (2.9-5.1) | 3.3 (3.0-4.9) | 0.71 |
AFP: Alpha-fetoprotein; ALT: Alanine aminotransferase; AP: Alkaline Phosphatase; AST: Aspartate aminotransferase; γGT: Gamma glutamil transpeptidase; INR: International Normatized Ratio; NASH: Non-alcoholic steatohepatitis.
Microbiome diversity is just one factor to consider when analyzing any ecosystem along with its stability, structure and function [1–5]. Many studies have used diversity as a key measure in their analysis, often assuming that diversity correlates with a “healthy” one. Diversity can be assessed by two specific methods, alpha diversity and beta diversity.
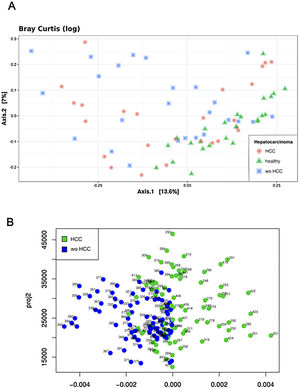
The Shannon's alpha diversity index indicated that healthy subjects showed the most diverse dataset of gut microbiome compared to cirrhotic cases and controls (ANOVA; P<0.05). The HCC dataset was less diverse than healthy controls, but still more diverse than the wo-HCC dataset (Tukey's honest significance test; P<0.05). Significant differences were found in the healthy vs HCC vs wo-HCC groups. There was not a clear differentiation between HCC and wo HCC groups in the PCoA and ADONIS results (Supplementary Fig. 1), so we performed a principal fitted components analysis with dimension reduction that showed a more precise and clear separation between groups (Supplementary Fig. 2).
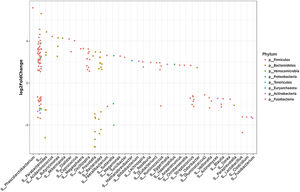
3.3Taxonomic distribution and differential abundance analysis of the gut microbiomeTaxonomic abundance at Phylum, Family and Genus levels were investigated for cases, wo-HCC and healthy groups. Of note there was expansion of non-beneficial taxa such as Bacteroidaceae and reduction of Prevotellaceae in the HCC group that was reflected at the genus level in Bacteriodes and Prevotella. Consequently, the bacteriodes/prevotella ratio was greater in HCC than in the two other groups (Fig. 1A).
 HCC, non-
HCC, non-Taxonomic abundance at phylum, family and genus levels between HCC, non-HCC patients and healthy individuals (Panel A) and differential abundance analysis using Phyloseq (Panel B). (A) A bacteriodes/prevotella ratio was greater in HCC than in non-HCC controls and healthy controls; (B) Several genera showed interesting correlations such as Fusobacterium, Prevotella, Streptoccocus, S24-7 (Phylum Bacteroidetes) and an unknown genus (phylum Firmicutes, family Leuconostocaceae), all of which were decreased 2 to 5-fold in HCC group. The ANCOM analysis identified 3 taxa to be associated with differences between cases and controls. Two OTU's from the Odoribacteraceae family, genus Odoribacter and Butyricimonas were also identified to be more differentialy abundant in HCC samples in the DESEQ2 analysis. The other OTU belong to the Lachnospiraceae family genus Dorea.
To further investigate which taxa accounted for the observed specific differences, we performed a differential abundance analysis (Fig. 1B). Several genera showed interesting correlations such as Fusobacterium, Prevotella, Streptococcus, S24-7 (Phylum Bacteroidetes) and an unknown genus (phylum Firmicutes, family Leuconostocaceae), all of which were statistically significant decreased, 2–5-fold, in HCC group. On the other hand, Haemophilus, Eggerthella, Bifidobacterium, Butyricimonas, Christensella, Odoribacter, an unknown genus phylum Tenericutes and an unknown genus, phylum Firmicutes, family Erysipelotrichaceae, were all elevated by 2–3-fold in HCC group (Fig. 1A and B).
Individual OTUs within some of the genera showed greater or lower values of fold change in the HCC group. For instance, several OTUs in Prevotella genus decreased between 3 and 6-fold but other Prevotella OTUs increased 1.5–3-fold. The mean fold change for genus Prevotella indicated a significant 3-fold decreased as shown in Fig. 1A and B. A full panorama involving the complete dataset of OTUs analyzed with significant changes is observed in Supplementary Fig. 3. Interestingly, all the changes in abundance in Fig. 2A and B correlated with an increase in the predicted metabolism of NLR signaling pathway in the HCC group (Fig. 2A) and a trend toward higher IL-6 and TNF-α levels (Fig. 2B) showing a link between this microbiome changes and inflammatory pathways among patients with HCC.
 HCC and non-
HCC and non-NOD-like receptor signaling pathway in the HCC and non-HCC groups (Panels A and B). Interestingly, all the changes in abundance in HCC patients correlated with an increase in the predicted metabolism of NOD-like receptor signaling pathway (NLR). The NLR signaling pathway was previously reported to be involved in inflammatory and autoinflammatory processes (Panel B). HCC: Hepatocellular carcinoma.
To further explore which taxa were specifically associated with HCC, the ANCOM analysis identified 3 taxa associated with differences in the composition of gut microbiome between cases and controls (Fig. 3A). Two OTU's from the Odoribacteraceae family, genus Odoribacter and Butyricimonas (OTU's ID: X4454586; X988375), were also identified to be differentially abundant in HCC samples in the DESEQ2 analysis. The other OTU with a decreased abundance in the HCC individuals belonged to the Lachnospiraceae family genus Dorea (OTU's ID: X310608).
 HCC group predictor was performed. OTUs: Operational taxonomic units;
HCC group predictor was performed. OTUs: Operational taxonomic units; The ANCOM analysis for identifying OTUs (Panel A) and mean decrease in Gini index for the top ten predictor OTUs (Panel B). (A) OTUs found in the ANCOM analysis (abundance is log transformed) identified 3 taxa to be associated with differences between cases and controls from the Odoribacteraceae family, genus Odoribacter and Butyricimonas (OTU's ID: X4454586; X988375). The other OTU belongs to the Lachnospiraceae family genus Dorea (OTU's ID: X310608); (B) An analysis using OTU's relative abundance as HCC group predictor was performed. OTUs: Operational taxonomic units; HCC: Hepatocellular carcinoma.
These 3 OTUs that were identify in patients with HCC were included in a predictive model to identify and classify HCC samples among controls w-HCC and healthy subjects. We developed a Random Forest classifier using these OTU's relative abundance as HCC group predictor (Fig. 3B). Then we selected these three OTU's as features and trained a new model. This resulted in a model with an improved performance, in which the testing classification accuracy had an AUROC of 0.75 (95% CI: 0.51–0.91), with a sensitivity of 0.80, specificity of 0.70, positive predictive value of 0.72 and negative predictive value of 0.78; with a positive and negative likelihood ratios of 2.66 and 0.28, respectively.
3.5Sensitivity analysisWe further performed a sensitivity analysis excluding patients under rifaximin (n=8) and lactulose (n=8). We found that there were no significant differences regarding microbiome profile between cases and controls after exclusion of this important confounder factors.
4DiscussionTo the best of our knowledge, this is one of two unique observational studies to determine the gut microbiome profile among cirrhotic patients with and without HCC [28,29]. Patients with HCC showed a more diverse gut microbiome than wo-HCC group. First, patients with HCC had specific changes in family members of Firmicutes including a 3-fold increased in abundance of Erysipelotrichaceae and a 5-fold decreased in family Leuconostocaceae when compared to controls. Second, genus Fusobacterium was found to be 5-fold lower in abundance in HCC vs wo-HCC. Third, the ratio bacteriodes/prevotella was increased in HCC cases due to the significant decrease in the genus prevotella. This pattern has been associated with an inflammatory milieu with increased activation of NLR signaling pathways. Finally, three OTU's were identified as potential biomarkers of HCC, including Odoribacteraceae family, genus Odoribacter and Butyricimonas that showed a higher abundance, while a decreased abundance was detected in Lachnospiraceae family genus Dorea in HCC subjects.
Microbiome diversity is just one factor to consider when analyzing any ecosystem along with its stability, structure and function [1–5]. Many studies have used diversity as a key measure in their analysis, often assuming that diversity correlates with a “healthy” one. This assumption might not be always the case. We observed that HCC patients had a less diversity index when compared to healthy controls and a more diverse index when compared to cirrhotic patients without HCC. This might underline the importance of a more complex microbial ecosystem including richness (number of different species in a community) and evenness (relative abundance).
Previous observational and small studies, as the present one, have found some changes in the gut microbiome of cirrhosis including a decreased abundance of Lachnospiraceae, Ruminococcaceae and Clostridialies and a higher abundance of Enterobacteriaceae and Bacteriodaceae [1]. Infection or inflammation triggers changes in gut microbiome with a relative increase in Enterobacteriaceae and Bacteriodaceae [2–5]. In our study, a higher abundance of Enterobacteriaceae was observed in all cirrhotic patients. Recent studies have shown that probiotics decrease inflammatory mediators including tumor necrosis factor (TNF-α) and endotoxemia [21]. However, this microbiome profile in cirrhosis might be unchanged after the eradication of the injurious factor that originated the liver disease as it was shown in patients after HCV eradication [21].
Although inflammation and tumorigenesis have been associated with HCC and cirrhosis, a link between changes in gut microbiome, inflammation and liver cancer has only been reported in murine models [9,10]. In our study, the HCC group showed an expansion of certain families or genera of microbes associated with an inflammatory milieu. These findings suggest a tumor specific expansion of microbiota [22].
It is interesting to note that some families in the phylum Firmicutes were either significantly elevated or decreased in HCC cases vs controls wo-HCC. We observed a 3-fold increased in abundance of Erysipelotrichaceae in HCC patients. This family of firmicutes has been implicated in inflammation related to gastrointestinal diseases and in colorectal cancer [22]. On the other hand, family Leuconostocaceae, a main producer of acetate and lactate, was decreased by 5-fold in HCC cases vs controls wo-HCC, as also observed in ulcerative colitis [23]. Moreover, several members of the gut microbiome were also found significantly decreased in HCC cases. The genus Fusobacterium, associated with tumorigenesis process and a proinflammatory microenvironment in the gut [22], was decreased by 5-fold in HCC patients showing that a specific microbiome related-cancer might be found in different tumors. Bacteroides and Prevotella are major genus of the Bacteriodetes phylum present in the gut and are mutually excluded in the dominance. Prevotella was also linked to chronic inflammation processes. In our study, the ratio bacteriodes/prevotella was increased in HCC cases due to the significant decrease in the genus prevotella and no significant changes in the genus bacteroides. In fact, prevotella was decreased by at least 3-fold in HCC cases.
Consequently, it seems that in cirrhosis, inflammation and tumorigenesis in HCC might be closely related [9,10]. It has been previously observed a “leaky” gut accounting for an impaired intestinal barrier function and bacterial overgrowth leads to the development of main clinical events in cirrhosis. These bacterial products induce inflammation through activation of TLR and NLRs in the liver, inducing a inflammatory environment of cancer development [9,10]. NOD-like receptors are intracellular sensors with central roles in innate and adaptive immunity [20]. In our study, changes in abundance in HCC patients correlated with an increase in the predicted metabolism of NOD-like receptor (NLRs) signaling pathway. In the study protocol we hypothesized that HCC might be an inflammatory cancer as was previously reported [9–11]. So, we included measurement of two important inflammatory cytokines. In a subsequent functional analysis considering the type of microbiome found, we observed that these findings were associated with inflammatory pathways (NLR). Although there was not a significant difference observed in cytokines levels, there was a trend toward a pro-inflammatory profile. Probably these differences did not achieve statistical significance mainly because of a beta error considering a low sample size. However, we consider this data of relevance in the context of our in silico functional analysis that linked the microbiome profile among HCC patients with NLR pathways.
The key question is whether these findings can be translated into clinical relevant knowledge in the field of HCC pathogenesis or a specific biomarker. Grat and colleagues have previously found in a study, without analyzing specific microbiota with third generation sequencing such as Illumina, that the number of colony-forming-units of E Coli were higher in HCC patients. That study included 15 patients with and 15 without HCC who were waiting for a liver transplantation. No functional analysis and no cytokine measurement were made but the authors concluded that this profile of intestinal overgrowth may contribute to HCC development [29]. Another recently published study in parallel to ours reports similar findings [28]. They perform a comparative and descriptive analysis among non-alcoholic liver cirrhosis patients, with and without HCC (n=21/20), and 20 healthy subjects [28]. They found that patients with HCC and NAFLD had higher levels of inflammatory cytokines related with a higher abundance of non-beneficial taxa such as Enterobacteriaceae and a reduction in beneficial taxa such as Akkermansia. Bacteroides and Ruminococcaceae were increased in HCC group [28]. This study was specifically done in NAFLD patients whereas our study included a small number of patients with this etiology. We did not find any significant inter-group differences considering different etiologies. Among cases–controls, both were well matched and no significant differences were observed regarding type of etiology. This issue should be further explored. We found 3 OTUs that were identified in HCC patients and tried to validate our findings in a random subset of our HCC/non-HCC patients. We performed a machine learning analysis by the construction of random forest models in order to predict any group from differences in abundance species. Three different OTU's, 2 from the Odoribacteraceae family, genus Odoribacter and Butyricimonas were identified to be differentially abundant in HCC samples. Interestingly, the genus Odoribacter has been linked and associated with colon cancer tumorigenesis [24]. In order to validate the use of these 3 OTUs to identify and discriminate patients with HCC from those without HCC, we performed a similar technique to bootstrapping, exposing these 3 OUT's in 1000 samples.
Another sensitivity analysis excluding patients under lactulose or rifaximin was performed, as these drugs could have been potential confounder factors. There were no significant differences in the proportion of cases vs controls under these drugs and in the microbiome profile between cases and controls (without HCC) after excluding patients with lactulose and rifaximine.
We recognize that this study has limitations. First, although case-control studies have several biases, a strict revision of selection and information bias was assessed. These preliminary observations could justify a longitudinal assessment of gut microbiota in cirrhotic patients and evaluate if this type of microbiome could be a new biomarker of HCC. However, these findings might not be applicable to all HCC patients, given the fact that there might have been other co-factors inducing inter-group and within-group variability. The fact that we were still able to find 3 OTUs that were significantly different in this small cohort without completely controlling for diet is interesting and remarkable [25]. Moreover, there is conflicting data regarding cancer development with these interventions [26]. Our results might be in line with previous observations in other research studies regarding the effect of microbiota in cancer development or growth, thus opening a new debate whether the “gut-liver axis” and its inflammatory pathways promotes liver cancer development [9,10,27].
In conclusion, we found several different genera and families in the gut microbiota that were decreased or elevated in HCC cases vs controls (wo-HCC). Patients with HCC had a 3-fold increase of Erysipelotrichaceae and a 5-fold decrease in Leuconostocaceae family, a 5-fold decreased abundance in genus Fusobacterium and an increased ratio bacteriodes/prevotella when compared to cirrhotic patients without HCC. This pattern was associated with inflammatory pathways. Three OTU's were identified as potential biomarkers of HCC, including a highly abundant genus Odoribacter and Butyricimonas with a decreased abundance in genus Dorea. These findings should be further explored as a novel biomarker of HCC development in larger cohort studies and eventually, in an intervention prevention trial.
Author's contributionsConcept and design, statistical analysis, writing of article by Piñero F, Vazquez M, Rohr C, Bare P and Fay F; Data recording, critical review of the manuscript by Mendizabal M and Sciara M; Cytokine serum analysis: Bare P; Statistical analysis from Piñero F and Rohr C. Senior author, guarantor and mentor of this research: Silva M.
Financial supportThis research received a specific grant from the National Institute of Cancer (INC), Argentina.
Conflicts of interestThe authors of this manuscript have no conflicts of interest to disclose as described by Annals of Hepatology. All authors have no conflicts of interest to report.
We thank the National Institute of Cancer (INC), Buenos Aires, Argentina (No. ID-190) for supporting this research.