



Reactive oxygen species (ROS) act as signaling intermediates regulting multiple cellular processes. The fate and disposal of the signaling species are determined by the actions of antioxidants, particularly glutathione (GSH). The mitochondrial pool of GSH (mGSH) arises from the transport of cytosol GSH by a specific mitochondrial carrier and is responsible for the maintenance of a healthy competent organelle. The depletion of mGSH upon impairment of the mitochondrial transport activity leaves mitochondria unprotected from damaging effects of ROS overgeneration within the mitochondrial electron transport chain. Tumor necrosis factor-α (TNF-α) has emerged as a key player in the progression of the alcohol-induced liver disease (ALD), and is known to target mitochondria. Key components of TNF signaling include sphingolipids, particularly ceramide generated from acidic sphingomyelinase activation serving as a source for gangliosides. In experimental models alcohol consumption enhances cholesterol levels and subsequent deposition into mitochondria resulting in selective decrease in the mGSH stores which is sufficient by itself to sensitize hepatocytes to TNF-α-mediated cell death. Thus, the combination of TNF-α overproduction, enhanced glycosphingolipid generation and selective mGSH depletion by alcohol intake cooperate making the liver sensitive to alcohol.
Oxidative stress is generally considered the result of an imbalance between two opposing, antagonistic forces, free radicals and antioxidants, in which the effects of the former predominate over the compensating action of the latter. Free radicals are molecules or fragments of molecules containing unpaired electron in their outermost orbitals. Because electrons tend to pair, these unpaired electrons intend to look for a partner. Therefore, most free radicals are extremely reactive and as a result, short-lived. Some stable molecules (e.g. nitric oxide) contain unpaired electrons in their orbitals and may be regarded as free radicals. When these unpaired electrons are centered on oxygen, these derivatives are termed reactive oxygen species (ROS). An imbalance between ROS and antioxidants can be accomplished by excessive generation of ROS and/or limited availability of antioxidants.
Glutathione (GSH), the most abundant non-protein thiol in cells, plays a key role in the disposition of ROS. In this review I will focus on the specific role of mitochondrial GSH pool (mGSH), which constitutes aproximately 10-15% of the cellular GSH content and gets there against an unfavourable electrochemical gradient. Particularly, I will summarize the evidence for the role of mGSH in the progression of alcohol-induced liver disease (ALD) and how mGSH depletion sensitizes hepatocytes against tumor-necrosis factor-mediated cell death.
ALD and TNFCirrhosis is the culmination of alcoholic liver disease (ALD) and is still one of the leading causes of death in developed countries. Despite intense research, the pathogenesis of ALD is not yet completely known and this lack of understanding limits the efficacy of treatment. In addition to the potential contributory role of malnutrition in the progression of the disease, the oxidative metabolism of alcohol within the liver generates a series of molecular intermediates that set in motion cellular and molecular cascades that contribute to the dysfunction of the liver. Thus, while the chronic metabolism of alcohol may perturb the appropriate redox potential of the hepatocyte due to the limited renovation of NAD+ equivalents, the constant formation of acetaldehyde, an extremely reactive alcohol by-product, contributes to the functional and structural alterations of the liver.1 The combined action of these factors derived from the biotransformation of alcohol unbalance the equilibrium between antioxidants and ROS generation, leading to oxidative stress, considered a major factor in the pathogenesis of the disease.2
Elevated cytokine levels cause symptoms of chronic inflammation. The liver also plays a role in the inflammatory response, both as a potential site of chronic inflammatory diseases as in ALD, and as a source of phagocytes and cytokines. Dysregulated TNF metabolism in ALD was first described more than a decade ago with the observation that cultured monocytes obtained from patients with alcoholic hepatitits spontaneously produced more TNF either under basal conditions and particularly upon LPS challenge.3,4 The relevance of these pioneering observations in patients has been recognized in animal models, particularly in TNFR-1 knockout mice5 in which alcohol-induced liver damage was diminished compared to wild type mice. Moreover, depletion of Kupffer cells, a major source of TNF and other cytokines, prevents liver injury. Thus, the release of TNF overproduction by monocytes and Kupffer cells and its effects on hepatocytes will impact on the sensitization of the liver to alcohol.6 Since TNF is such an important pathogenetic factor in the disease, the characterization of its signaling pathway would be instrumental for the management of ALD. In addition to the issue of quantity (TNF overgeneration by alcohol consumption), it is also conceivable that alcohol may actually induce in hepatocytes an aberrant response to TNF. Normally, hepatocytes are resistant to TNF and to overcome this resistance survival pathways ought to be blocked to unmask the full cytotoxic potential of TNF. Intriguingly, hepatocytes isolated from alcohol-fed rats develop an unusual sensitivity to TNF exposure in the absence of any other sensitizing factor.7,8 Since parenchymal cells constitute at least two thirds of the total cell population of the liver the susceptibility of hepatocytes to TNF by alcohol may be devastating for the organ. Uncovering the molecular factors underlying this phenomena may be of clinical relevance in the treatment of the disease allowing the design of therapeutic strategies to neutralize specific TNF intermediates signaling entities or to increase the mGSH content.
Glycosphingolipids in TNF signalingThe cellular signaling network used by TNF to transmit its effects to the cell interior is complex and involves generation of a wide variety of intermediates and proteinprotein interactions.9 TNF-induced cell death is mediated by its binding to TNFR-1, causing receptor oligomerization and subsequent recruitment of the adapter protein TRADD. The binding of TRADD to FADD activates caspase-8, which, in turn, cuts specific target proteins including pro-caspase-3. Furthermore, the binding of TRADD to the downstream transducer protein TRAF-2 and the protein kinase NIK, results in the activation of transcription factor NF-KB, which activates the transcription of antiapoptotic genes, including inhibitors of apoptosis proteins (cIAPs), that antagonize the FADD-mediated death signals.9 TNF has also been reported to generate sphingolipids that have been implicated as effectors of TNF-mediated apoptosis.10,11 Ceramide has attracted considerable attention due to its involvement in the modulation of cell death, although it also functions in the regulation of many other cellular processes, e.g. differentiation, proliferation, gene regulation. Ceramide levels can increase through several mechanisms, including the de novo synthesis from L-serine and palmitoyl-CoA catalyzed by the action of serine-palmitoyl transferase and the activation of ceramide synthase. On the other hand, ceramide levels can increase by hydrolysis of membrane sphingomyelin by the action of sphingomyelinases (SMases). This pathway may be of significance in promoting specific macrodomain formation in the plasma membrane allowing oligomerization of certain cell surface proteins such as ligated receptors (TNF family).12 Several sphin-gomyelinases have been identified, although not all of them have been fully characterized. The magnesium-dependent, membrane-bound, neutral sphingomyelinase (NSMase) has been linked to apoptosis mediated by chemotherapeutic agents, serum starvation, TNF, and Fas. Acidic sphingomyelinase (ASMase), which displays an optimum pH around 4.8, has been further subclassified into the endosomal/lysosomal ASMase and a secretory Zn2+-dependent SMase. Studies in non-parenchymal cells provided indirect evidence for a role of NSMase in TNF-induced cell death as fibroblasts deficient in FAN were resistant to TNF.13 On the other hand, ASMase has been shown to contribute to developmental apoptosis.14 In addition, hepatocytes deficient in ASMase showed resistance to Fas-and radiation-mediated apoptosis.15,16 Our recent data indicate a contributory role for ASMase in TNF-mediated hepatocellular apoptosis and liver damage.17 Thus, based on these findings our current hypothesis establishes that ASMase may play a pathogenic role in the pathology of ALD acting as an intermediate of TNF, and its specific role in the progression of the disease is currently under investigation.
In addition to the evidence for a direct role for ceramide in the regulation of stress induced cell death, ceramide provides the carbon backbone for the synthesis of complex glycosphingolipids (GSLs), such as gangliosides. GLSs are synthesized from their precursor glucosylceramide upon the addition of a glucose residue to ceramide catalyzed by the glucosylceramide synthase, a resident enzyme located within the Golgi network. The synthesis is coupled to the exocytotic vesicle flow through the Golgi apparatus to the their predominant location in the outer leaflet of plasma membrane where they play important regulatory roles in signal transduction, proliferation and cell adhesion.18 In particular, ganglioside GD3 (GD3), a sialic acid-containing GSLs, has attracted considerable attention due to its emerging role as a cell death effector, activating the mitochondrial-dependent apoptosome through sequential The mitochondrial permeability transition (MPT) induction, cytochrome c release and caspase activation. As with ceramide, the cellular levels of GD3 increase in response to Fas or TNF,19,20 whereas the down regulation of GD3 synthase, the enyzme responsible for GD3 synthesis from its precursor GM3, prevents Fas-TNF-or β-amyloid-induced cell death.19-22 While most of the support for the apoptotic function of GD3 has derived from in vitro studies with isolated mitochondria, recent data monitored the distribution and targeting of GD3 to mitochondria in response to TNF.23 Furthermore, the inhibition of glucosylceramide synthase protected cultured rat hepatocytes from TNF-induced apoptosis with the downregulation of GSLs and enhancement of ceramide,17 indicating the involvement of GSLs. Moreover, the downregulation of targeted GD3 synthase by antisense expressing vectors has been recently reported to protect human colon HT-29 cells to TNF-mediated cell death.21 Thus, these data provide strong evidence for the role of GSLs, in particular GD3, in the apoptotic signaling of TNF, and consequently its specific contribution to ALD is under investigation in our lab.
mGSH and ALDMitochondria have attracted considerable attention because of their role in cell death.24 Despite its universal presence in cells, GSH is not homogeneously distributed within cellular organelles. Most of the GSH is found in cytosol, comprising 80-85% of total cellular content, while, a 10-15% of the cellular pool is found in mitochondria where it reaches a concentration similar to that of cytosol.25 The mGSH pool originates from a specific transport system located in the inner mitochondrial membrane that translocates GSH from the cytosol into the mitochondrial matrix.26,27 Such a carrier operates efficiently in providing negatively charged GSH to the mitochondrial matrix, functioning against a steep electrical gradient imposed by the mitochondrial membrane potential.
Our studies in alcohol-fed rats (Lieber-DeCarli) revealed a selective depletion (~45-60%) in the mitochondrial pool of GSH.2 Alcohol intake impairs the cytosol to mitochondrial GSH transport of GSH as demonstrated in in vitro and in vivo tracer kinetic studies.28 This defect occurs preferentially in the mitochondria of the perivenous population of liver cells, precedes mitochondrial dysfunction25 and sensitizes isolated hepatocytes to exogenous oxidative stress-and cytokine-induced cell death.7,8,28 The depletion of mGSH by alcohol consumption was reproduced in the intragastric alcohol infusion model, preceding signs of alcohol-induced liver damage.29 In agreement with these findings it has recently been shown that intragastric alcohol feeding to rats resulted in a predominant mGSH depletion.30 The susceptibility of alcohol-fed rat derived hepatocytes to TNF-induced damage was reversed by mGSH repletion or antioxidants.7 An interesting report indicated that the selective depletion of mGSH induced by chronic alcohol consumption enhances the toxicity of acetaminophen.31 Thus, these observations point to a vital role of mGSH against cytotoxic insults, including TNF or acetaminophen, whose mechanisms of damage are dependent on mitochondria. In particular, since mitochondria generate important quantities of ROS, the limitation of mGSH by alcohol would favor the mitochondrial stimulation of ROS. Interestingly, because recent findings have indicated that superoxide anion acts on the matrix side of the mitochondrial inner membrane,32 and hence the depletion of GSH in the matrix would result in overproduction of hydrogen peroxide formed from superoxide could either harm vital mitochondrial components and/or favor cytotoxic mechanisms.
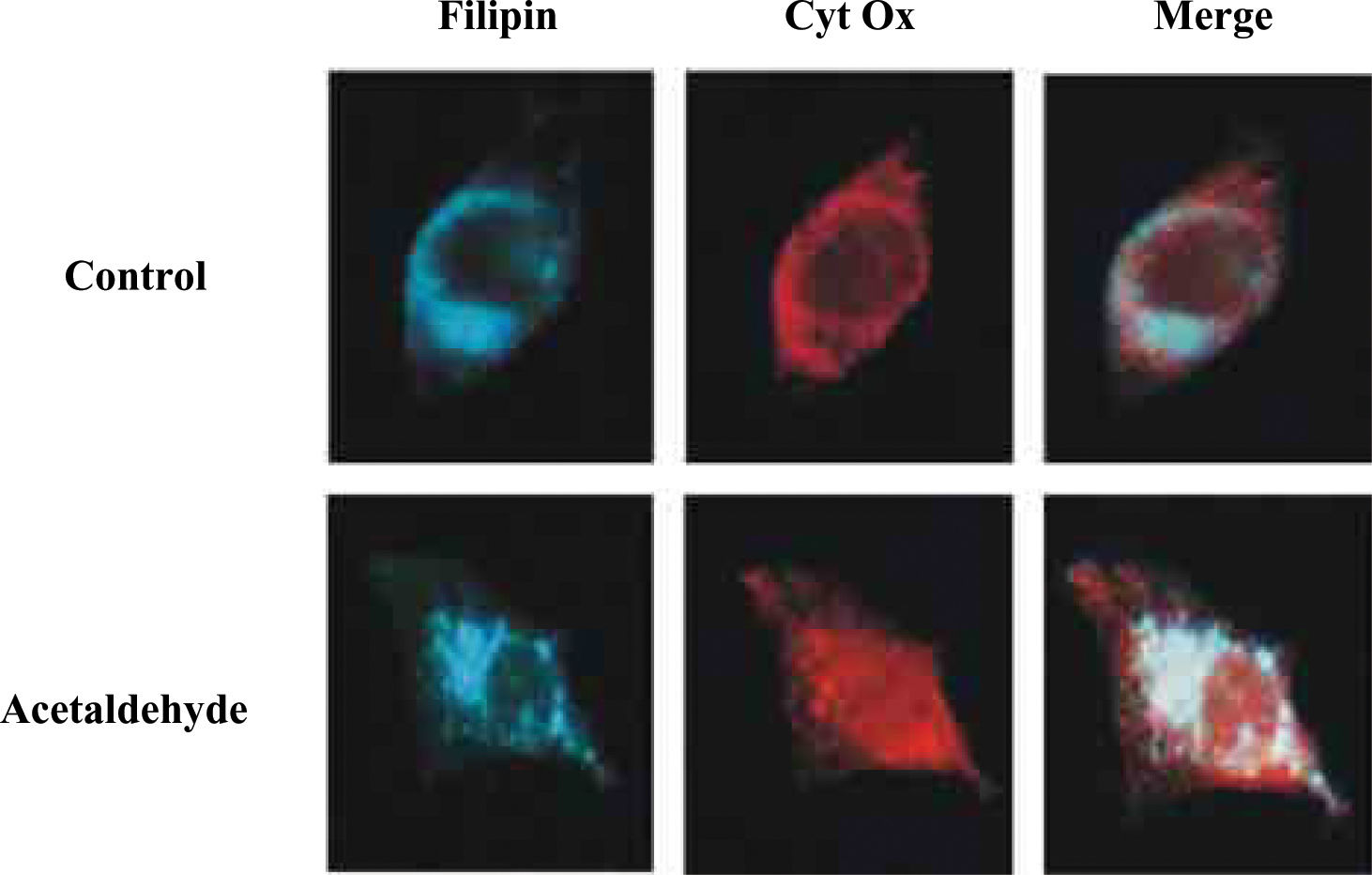
An important step towards the understanding of mGSH regulation by alcohol was its dependence on appropriate mitochondrial membrane fluidity range. Studies in intact mitochondria and mitoplasts from alcohol-fed rat liver indicated that the normalization of mitochondrial inner membrane fluidity in vitro by the fluidizing agent A2C, or in vivo by SAM or tauroursode-oxycholic acid (TUDCA), restores the kinetics of the mGSH carrier replenishing mGSH levels.7,27,33 Findings with mitochondria and mitoplasts showed that chronic alcohol enhanced the levels of total cholesterol, and that the fluidizing effect of SAM was mediated, in part, through reduction in the cholesterol/phospholipid molar ratio.27 Recently, we have investigated the role of acetaldehyde in the regulation of mGSH. These findigs underscore a contributory role of acetaldehyde in the impairment of mGSH transport induced by alcohol, as the incubation of HepG2 with acetaldehyde reproduced the effects elicited by alcohol.34 An interesting aspect of these findings was the description that acetaldehyde through endoplasmic reticulum stress stimulated the de novo cholesterol synthesis, its deposition in mitochondria resulting in increased membrane microviscosity and impaired transport of GSH into the mitochondria. Indeed, as shown in Figure 1 when the cholesterol content of HepG2 cells was examined in relationship with mitochondria by confocal microscopy we observed that acetaldehyde treatment induced the colocalization of free cholesterol probed by filipin with mitochondria. These findings with HepG2 cells were confirmed in isolated rat liver mitochondria enriched in free cholesterol. Consequently, through these sequence of events acetaldehyde sensitizes HepG2 cells to TNF, an effect prevented by cyclosporin A, GSH ethyl ester and lovastatin.34 Thus, due to the relevance of cholesterol deposition in mitochondria in the perturbarion of mGSH and subsequent sensitization to alcohol-mediated damage, the trafficking of cholesterol into the mitochondrial inner membrane may be of relevance for ALD. In this regard specific StAR-related lipid transfer proteins35 may be induced or activated by chronic alcohol intake in hepatocytes. If confirmed these proteins may emerge as new therapeutic targets for ALD management.
. Cells were washed and fixed prior to permeabilization with saponin and incubation with anti-human cytochrome oxidase subunit II (Molecular probes) and incubated with Cy3-conjugated secondary antibody anti-mouse F(ab’)2 (Jackson ImmunoResearch Labs) for 60min. Cholesterol was stained with filipin (20μg/ml) together with the secondary antibody. Samples were washed and chambersli des mounted with Mowiol and viewed with a Leica DMR confocal microcope.")
Localization of cholesterol and mitochondria in response to acetaldehyde. HepG2 cells were cultured in the presence of acetaldehyde (100μM for 3 days). Cells were washed and fixed prior to permeabilization with saponin and incubation with anti-human cytochrome oxidase subunit II (Molecular probes) and incubated with Cy3-conjugated secondary antibody anti-mouse F(ab’)2 (Jackson ImmunoResearch Labs) for 60min. Cholesterol was stained with filipin (20μg/ml) together with the secondary antibody. Samples were washed and chambersli des mounted with Mowiol and viewed with a Leica DMR confocal microcope.
Early studies showed that mitochondria are devoid of the enzymatic machinery to synthesize GSH from its constituent aminoacids. The mGSH pool arises from the cytosol by a carrier-mediated transport to overcome its unfavourable entry against an electrochemical gradient across the mitochondrial inner membrane.26,36-39 Characterization of the transport of GSH into rat liver mitochondria indicated an active, energy-dependent process stimulated by ATP and inhibited by glutamate and protonophores (e.g. FCCP) that collapse the protonmotive force.26 Although, the molecular identity of the mGSH carrier has remained elusive,40 recent evidence from rabbit kidney mitochondria indicated a role for the dicarboxylate and 2-oxoglutarate carriers in the mitochondrial transport of GSH.37-39 Particularly, the reconstitution of a partially purified preparation of mitochondrial transporters from kidney mitoplasts in proteoliposomes and the overexpression in NRK-52E cells with a dicarboxylate cDNA provided further evidence for this mitochondrial carrier as a GSH transporting polypeptide.37 In contrast to this role for the dicarboxylate carrier, the activity of the 2-oxoglutarate carrier as a GSH transporter has remained undefined.
Using the combination of the functional expression in Xenopus laevis oocytes and the inhibitor specificity and kinetic analises in isolated rat liver mitochondria, it has recently been shown that the 2-oxoglutarate carrier functions as a GSH transporting polypeptide in rat liver mitochondria, thus contributing to its origin.41 However, divergence in the kinetics of 2 oxoglutarate carrier and GSH seems to suggest the existence of additional carrier yet remaining to be identified. The mitochondrial transport of 2-oxoglutarate exhibits a single Michaelis-Menten component with kinetic parameters in the range of those reported previously for renal mitochondria.39 However, the GSH transport reported in isolated mitochondria from rat liver,26,27 HepG2 cells34 or X laevis oo-cytes microinjected with total hepatic mRNA,42 characteristically display two kinetic components with high and low affinity for GSH transport. Intriguingly the kinetic parameters of the low affinity component of GSH transport26,27 and that of 2-oxoglutarate are similar both showing similar Vmax and a Km in the mM range. While this suggests that the 2-oxoglutarate carrier may be responsible for the low affinity transport of GSH in rat liver mitochondria, there may be alternative carrier(s) that account for the high affinity transport of GSH. The role of the recently cloned human mitochondrial oxodicarboxylate carriers Odcp1 and Odcp2,43,44 which mediate a counter-exchange between oxodicarboxylates on the transport of GSH in rat liver mitochondria remains to be explored.
Role of mGSH in the integrity of vital mitochondrial componentsThe mitochondrial permeability transition (MPT) involves a sudden and initially reversible increase to solutes with molecular mass up to 1,500 Da. The rapid change in permeability associated with transition causes mitochondrial depolarization, uncoupling of oxidative phosphorylation, release of intramitochondrial solutes, large amplitude swelling and outer membrane rupture.45 This phenomenon is mediated by opening of a large conductance channel, the permeability transition pore (PTP), located at the contact sites between mitochondrial membranes. Although the molecular identity of this multiprotein complex pore is only partially defined, some of its core components include the adenine nucleotide translocator (ANT), found in the inner membrane and the voltage-dependent anion channel (VDAC), located in the outermembrane. Other proteins believed to be involved are the peripheral benzodiazepine receptor (PBR), hexokinase II (a cytosolic protein), creatine kinase (located in the intermembrane space), cyclophilin D (located in the mitochondrial matrix) as well as Bax/Bcl-2-like proteins.46
The MPT has been shown to function as a key device in the control of cell survival regardless of the phenotype of cell death.24,45 Agents that induce or prevent MPT modulate cell survival. As a result of mitochondrial membrane permeabilization death-promoting factors are released from the mitochondria intermembrane space, thereby stimulating the execution phase of the cell death program. Such activators include certain inactive caspases precursors, cytochrome c which triggers the assembly of the cytochrome c/Apaf-1/pro-caspase-9 activation complex, the apoptosome;47,48 heat shock protein 10, which favors apoptosome activation;49 the apoptosis-inducing factor (AIF) and endonuclease G, both of which mediate the cleavage of DNA during caspase-independent cell death;50,51 Smac/DIABLO and Omi/HtrA that ensure inactivation of caspase inhibitors IAPs;52-54 and pre-processed caspase-9.55
Although one of the consequences derived from MPT is the rupture of the outer membrane, some studies have pointed out that outer membrane permeabilization may occur in the absence of disrupted inner membrane.56-59 Indeed there is growing evidence supporting a MPT-independent release of cyt C during apoptosis. Although it remains unclear exactly how mitochondrial outer-membrane permeabilization occurs either by MPT, large pore opening or even hyperpolarization subsequent to the closure of VDAC, it is clear that it represents a key event allowing the release of intermembrane proteins, e.g. cyt c, that initiate the apoptosome activation leading to cell death.
Different factors may modify the magnitude of mitochondrial response to the MPT including Ca2+ and ADP levels, matrix pH and ΔΨm, mitochondrial energetic status, lipid peroxidation, oxidative stress due to ROS overproduction and mGSH depletion.46 In pathological conditions, one or several of these MPT-regulating factors may contribute to altered susceptibility to MPT. Of relevance to ALD, it has been shown that liver mitochondria from alcohol-fed rats showed an increased susceptibility to MPT induced by different stimuli.60 The underlying mechanisms for the latter observations are not completely understood and may include changes induced by alcohol to MPT components (post-translational modifications), alterations in the lipid milieu and/or depletion of mGSH. Regardless of the mechanism leading to MPT it represents a key point in the control of cell death and thus the potentiation of MPT by alcohol may be essential in the pathogenesis of the disease. Thus, the characterization of the mechanisms underlying this process may be of significance for the treatment of the disease.
Concluding remarks and future directionsALD is a complex multifactorial process intiated from the oxidative metabolism of alcohol in the liver. The metabolic disturbances of this pathway converge at mitochondria which undergoes a selective depletion of mGSH pool defense subsequent to the accumulation of cholesterol leading to perturbation of the mitochondrial inner membrane fluidity. While this occurs within hepatocytes non-parenchymal cells are stimulated by alcohol to release inflammatory cytokines, e.g. TNF which act upon unprotected hepatocytes. The target of TNF on hepatocytes generates sphingolipid intermediates which upon their trafficking into mitochondria promote the release of proapoptotic factors into the cytosol of hepatocytes resulting in the building of the apoptosome that precedes the apoptotic destruction of the cell. As ROS accumulate in a self-fulfilling circle particularly with limited availability of mGSH, caspase may become inactivated switching cell death from apoptosis to necrosis. Thus, emerging from this picture a clear understanding on the regulation of mitochondrial lipid trafficking and TNF signaling acting on mitochondria may be essential to design a broad range of therapeutic avenues to rescue mGSH depleted hepatocytes against the prey of ROS overgeneration. A clear conclusion drawn from the evidenced observed in experimental models is that fat deposition may actually sensitize hepatocytes against compromising challenges introduced by alcohol metabolism including TNF overgeneration, mitochondrial dysfunction (ATP depletion) o hypoxia. These same ingredients may gather in nonalcoholic steatohepatitis, an increasingly recognized health problem, with many characteristic features indistinguishable from ALD.61 If the microvesicular fat storage containing free cholesterol deposits traffic to mitochondria may lead to mGSH upon perturbation of mitochondrial membrane fluidity even in the absence of alcohol intake. Although the particular status of mGSH has not been reported up to date in human subjects it may constitute an important therapeutic target in the management of ALD to make hepatocytes withstand diverse challenges elicited by the oxidative biotransformation of alcohol within the liver.
AcknowledgmentsThe work was supported by Research Center for Liver and Pancreatic Diseases, P50 AA11999 and grant 1R21 AA014135-01 funded by the U.S. National Institute on Alcohol Abuse and Alcoholism, Plan Nacinal de I+D grants SAF01-2118 and Redes Temáticas de Investigación Cooperative G03715 and Red de Centros C03/02 supported by the Instituto de Salud Carlos III.



