



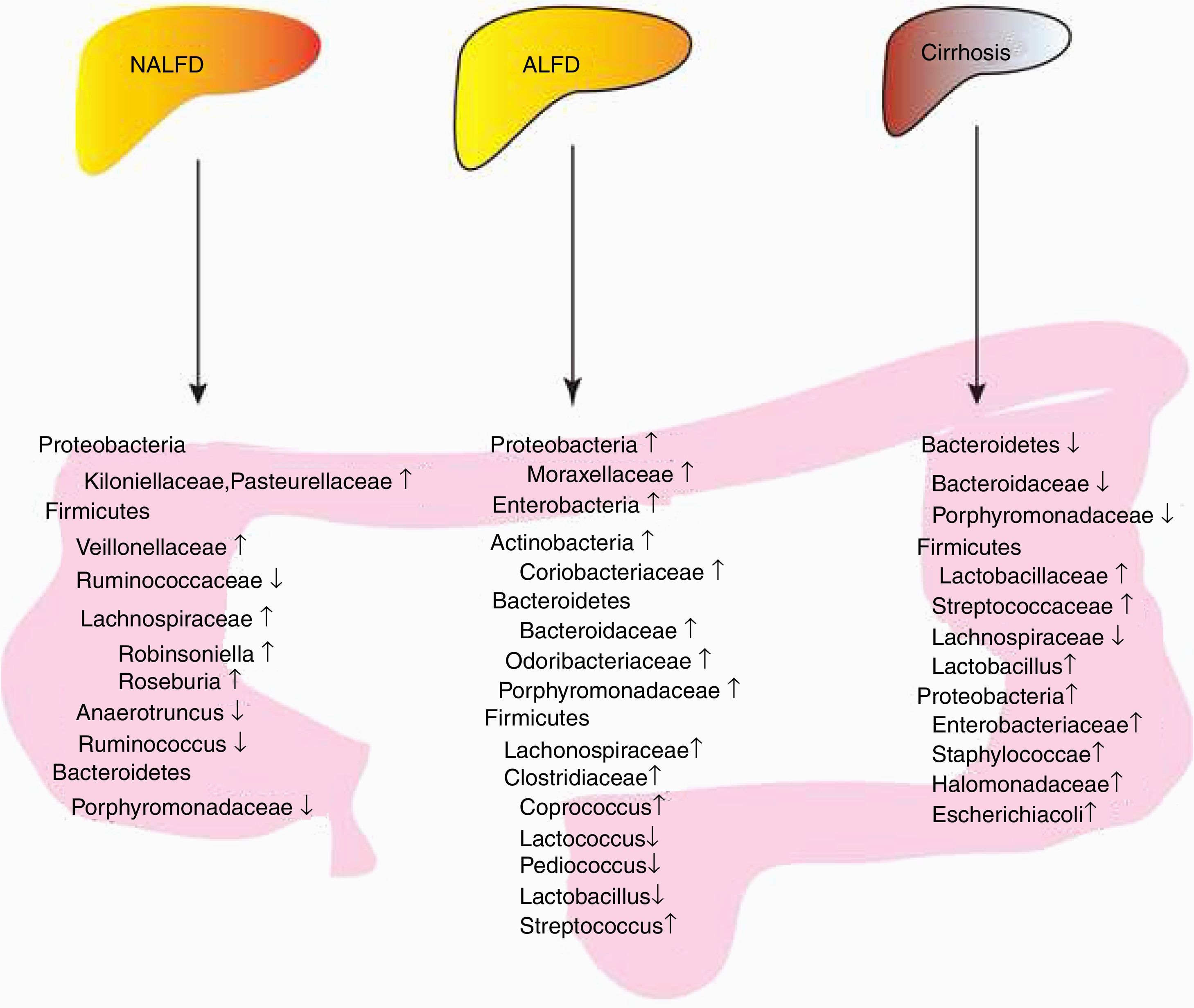
Non-alcoholic fatty liver disease (NAFLD) and alcoholic fatty liver disease (AFLD) are significant health burdens worldwide with a substantial rise in prevalence. Both can progress to liver cirrhosis. Recent studies have shown that the gut microbiome was associated with NAFLD/AFLD development and progression. The present review focuses on the characteristics of bacteria in NAFLD, AFLD and liver cirrhosis. The similarities and differences of intestinal bacteria are discussed.
This study reviews the existing literatures on the microbiota, fatty liver disease, and liver cirrhosis based on Pubmed database.
The study showed NAFLD was characterized by increased amounts of Lachnospiraceae from the phylum Firmicutes and Roseburia from the Lachnospiraceae family, and the proportion of Enterobacteria and Proteobacteria was increased after alcohol intake. Reduced Bacteroidetes was observed in cirrhosis. Microbiota can improve or aggravate the above liver diseases through several mechanisms, like increasing liver lipid metabolism, increasing alcohol production, increasing intestinal permeability, bacterial translocation, intestinal bacterial overgrowth, enteric dysbiosis, and impairing bile secretion.
Different hepatic diseases owned different intestinal bacterial characters. Microbiota can improve or aggravate three kinds of liver diseases through several mechanisms. However, the depletion of these bacteria is needed to verify their role in liver disease.
Apart from virus-induced liver disease, non-alcoholic fatty liver disease (NAFLD) and alcoholic fatty liver disease (AFLD) are the most common liver disorders in the world. Non-alcoholic steatohepatitis (NASH), one stage of NAFLD, and alcoholic steatohepatitis (ASH) can progress to cirrhosis and hepatocellular carcinoma (HCC), which are life-threatening [1]. Accumulating evidence has shown that intestinal bacteria play a key role in the development of NAFLD, regardless of the amounts of intestinal microbiota or colonic bacteria [2–5]. Bacteria might participate in the process of energy harvest, fermentation of polysaccharides, and regulation of bile-acid and choline metabolism [6]. In addition, acute-on-chronic alcohol feeding could change the intestinal bacteria, with increased Actinobacteria and decreased Verrucomicrobia/Akkermansia, the characteristics of which could influence liver inflammation, suggesting an important role of gut bacteria in the development of alcoholic liver disease (ALD) [7]. Furthermore, the administration of Akkermansia muciniphila could decrease liver injury and steatosis [8].
Despite studies that have shown that intestinal bacteria were related to NALFD and AFLD, the similarities and differences of intestinal bacteria profiles have not been well illustrated. Here, we reviewed the latest studies to identify specific bacteria associated with NAFLD, AFLD and cirrhosis and to provide a path to provide prevention and therapeutic strategies.
1.1Gut bacteria characteristics in NAFLDNon-alcoholic fatty liver disease (NAFLD) is regarded as a manifestation of the metabolic syndrome in liver, occurring in 80% of obese patients [9]. Studies have shown that NAFLD is associated with the gut microbiota. We will discuss the intestinal bacterial composition in NAFLD patients with or without obesity, and try to summarize the potential pathogenic bacteria leading to NAFLD.
In an animal study, C57BL/6J mice were fed a high-fat diet (HFD) freely for 16 weeks. Those who developed metabolic disorders, including steatosis, were regarded as responders, and those who did not were regarded as non-responders [10]. Then, the gut microbiota from responders and non-responders were transmitted to germ-free (GF) mice that were divided into a responder-receiver (RR) or non-responder-receiver (NRR) group. After 3 and 16 weeks of HFD (T3 and T16), at the genus level, RR mice had higher amounts of Barnesiella and Roseburia, while NRR mice had more Allobaculum [10]. At the phylum level, Firmicutes were represented more in RR mice at T3. At the species level, RR mice harbored more Lachnospiraceae bacteria 609 and Barnesiella intestinihominis at T3. Bacteroides vulgatus was higher in the NRR group. Their results demonstrated that gut microbiota affects systemic glucose homeostasis and liver lipid metabolism, indicating that gut microbiota can promote the occurrence and development of NAFLD [10]. Accumulating evidence from human studies also shows an association between NAFLD and intestinal bacteria. Gut microbiota from NASH patients (n=16) and healthy controls (n=22) were analyzed using 16S rRNA gene sequencing. The abundance of Faecalibacterium and Anaerosporobacter in feces of NASH patients was lower, while the abundance of accessory Parabacteroides and Allisonella was higher [11]. Thirty obese NAFLD patients and 30 controls were selected in a recent another clinical study. Stool analysis using multitag pyrosequencing demonstrated that obese NAFLD patients harbored increased amounts of Kiloniellaceae/Pasteurellaceae(belonging to Proteobacteria phylum), and Lactobacillaceae/Lachnospiraceae/Veillonellaceae (belonging to Firmicutes phylum) and decreased Ruminococcaceae (belonging to Firmicutes phylum) and Porphyromonadaceae (belonging to Bacteroidetes phylum) at the family level compared to controls [12]. At the genus level, obese NAFLD patients showed increased amounts of Lactobacillus (belonging to the Lactobacillaceae family) and Robinsoniella/Roseburia/Dorea (from Lachnospiraceae family) and lower amounts of Oscillibacter (of the Ruminococcaceae family) [12]. In another study, gut microbiota from obese patients without NASH, with NASH and healthy controls were analyzed using 16S rRNA sequencing [13]. The study showed that the only abundant phylum showing significant differences between obese patients and NASH patients was Proteobacteria [13], suggesting its dominant role in the development of NASH. At the family level, amounts of Enterobacteriaceae exhibited significant differences between NASH and obese patients. Their data suggest that microbiomes rich in ethanol-producing Escherichia may be a risk factor for the progression of disease from obesity to NASH [13]. Gut microbiota of lean-NALFD were examined in another study, where fecal samples from 43 lean-NAFLD and 83 healthy controls were collected and analyzed with 454 pyrosequencing of 16S rRNA [14]. The study demonstrated lower diversity and bacteria change at the phylum level in lean-NAFLD patients, with more Bacteroidetes and less Firmicutes [14]. Four families and 8 genera from Firmicutes, involving short-chain fatty acid-producing and 7α-dehydroxylating bacteria, were decreased in lean-NAFLD, including Lachnospiraceae, Ruminococcaceae, Lactobacillaceae and Peptostreptococcaceae [14]. Within Lachnospiraceae, Coprococcus and Anaerosporobacter were decreased in the lean-NAFLD group. In addition, lower proportions of Anaerotruncus and Ruminococcus from the Ruminococcaceae family and Lactobacillus of the Lactobacillaceae family were observed in the lean-NAFLD group [14]. Then, another study compared the gut microbiome from lean NASH patients versus overweight/obese patients with or without NASH, where fecal samples from13 biopsy-proven NASH patients and 10 controls were collected and analyzed with sequencing of 16S rRNA. There was no statistically significant difference in phylum level between the fecal bacteria of the obese, overweight and underweight NASH patients and the obese and underweight control group. However, at the level of bacterial genera, it showed that Faecalibacterium, Ruminococcus, Lactobacillus and Bifidobacterium abundance has significant difference in patients with NASH compared to control. In particular, decreased amounts of Faecalibacterium and Ruminococcus and Lactobacilli were found in patients with lean NASH compared with overweight/obese NASH. While NASH patients with obesity had rich amounts of Lactobacilli, overweight NASH patients with reduced Bifidobacterium [15]. Similarly, another children study showed that different gut microbiota were observed in obese children with or without NAFLD and healthy lean children using 16S rRNA sequencing. Compared to healthy lean and obese children, children with NAFLD had higher levels of Epsilonproteobacteria, GammaProteyia and Prevotella. There were more pathways of energy metabolism and lipid synthesis and alcohol production and fewer pathways of amino acid metabolism in NAFLD group, which would contribute to development of disease [16]. Gut microbiota profiling of pediatric non alcoholic fatty liver disease were examined in study, where fecal samples from 27 NAFLD, 26 NASH, or 8 obesity and 54 healthy controls were collected and analyzed with 454-Junior Genome Sequencer pyrosequencing of 16S rRNA. Compared to healthy controls, at the level of phylum, Actinobacteria abundance was increased in NAFLD patients, whereas Bacteroidetes abundance was decreased; at the level of family, in NAFLD patients Rikenellaceae were decreased; at the level of genus, in NAFLD patients Oscillospira were reduced and Ruminococcus,Blautia,Bradyrhizobium,Anaerococcus,Peptoniphilus,Propionibacterium acnes, Dorea increased. Compared to healthy controls, ethanol expression was also up-regulated in NASH patients [17]. It is well known that only a portion of patients with NAFLD develop fibrosis and then develop cirrhosis, gut microbiota from NASH (n=35) and no NASH (n=22) were analyzed using 16S rRNA gene sequencing. NASH and Significant fibrosis patients had increased levels of Bacteroides and reduced proportions of Prevotella. Ruminococcus abundance was significantly increased in Significant fibrosis patients [18]. In another study, gut microbiome compositions were analyzed by whole-genome shotgun sequencing of DNA method in order to distinguish mild/moderate NAFLD (n=72) and aggravated fibrosis (n=14). At the phylum level, Firmicutes was more common in mild/moderate NAFLD, while Proteobacteria was more common in aggravated fibrosis. At the species level, Bacteroides vulgates and Eubacterium rectal was more common in mild/moderate NAFLD, while Bacteroides vulgates and Escherichia coli was more common in aggravated fibrosis. Compared with mild/moderate NAFLD, the abundance of Ruminococcus obeum and Eubacterium rectale decreased significantly in aggravated fibrosis [19].
In conclusion, patients with NALFD had increased amounts of Lachnospiraceae/Veillonellaceae from the phylum Firmicutes and Kiloniellaceae/Pasteurellaceae from the phylum Proteobacteria in NAFLD, decreased amounts of Porphyromonadaceae from the phylum Bacteroidetes and Ruminococcaceae from the phylum Firmicutes (Fig. 1, Table 1).

Summary of bacterial changes in three liver diseases. Proteobacteria, Firmicutes and Bacterioidetes were dominated in patients with NALFD; Microbiota can improve or aggravate NAFLD through several mechanisms, including glucose homeostasis disorder, increased liver lipid metabolism, increased intestinal alcohol production and increased energy metabolism. Increased Enterobacteria, Proteobacteria and Bacteroidetes were observed in AFLD, while different genus from Firmicutes showed different levels;Microbiota can improve or aggravate ALFD through several mechanisms, including increased intestinal permeability, bacterial translocation, elevated plasma endotoxin, elevated fecal pH, intestinal bacterial overgrowth, enteric dysbiosis, and gut-barrier function destruction. A reduction of Bacteroidetes and increase of Proteobacteria was found in cirrhosis patients or animal models. And for Firmicutes, Lactobacillaceae and Stretococcaceae increased, while Lachnospiraceae and Lactobacillus decreased, Liver cirrhosis is associated with bile secretion disorders.
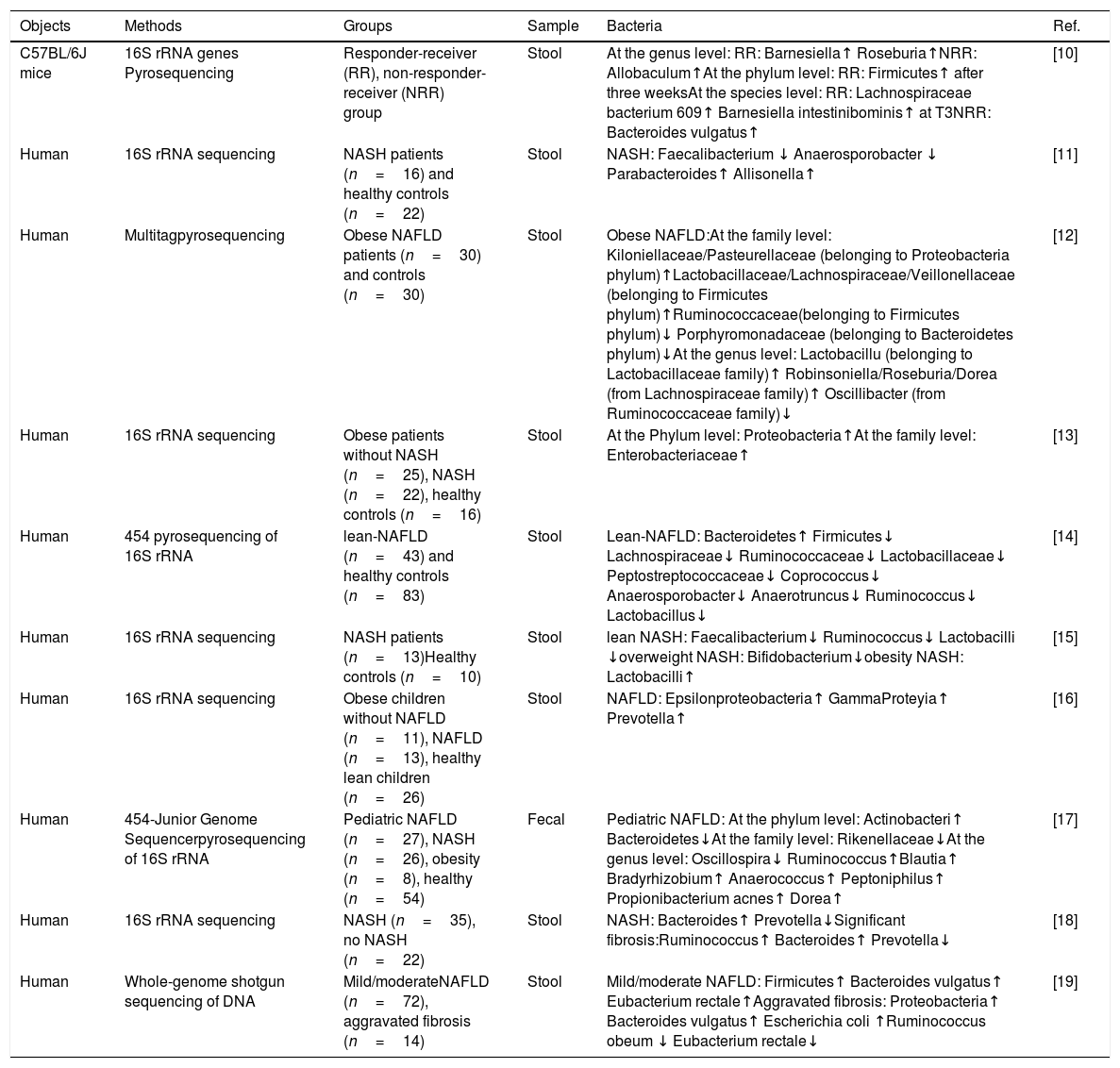
Characteristics of stool bacteria in NAFLD.
| Objects | Methods | Groups | Sample | Bacteria | Ref. |
|---|---|---|---|---|---|
| C57BL/6J mice | 16S rRNA genes Pyrosequencing | Responder-receiver (RR), non-responder-receiver (NRR) group | Stool | At the genus level: RR: Barnesiella↑ Roseburia↑NRR: Allobaculum↑At the phylum level: RR: Firmicutes↑ after three weeksAt the species level: RR: Lachnospiraceae bacterium 609↑ Barnesiella intestinibominis↑ at T3NRR: Bacteroides vulgatus↑ | [10] |
| Human | 16S rRNA sequencing | NASH patients (n=16) and healthy controls (n=22) | Stool | NASH: Faecalibacterium ↓ Anaerosporobacter ↓ Parabacteroides↑ Allisonella↑ | [11] |
| Human | Multitagpyrosequencing | Obese NAFLD patients (n=30) and controls (n=30) | Stool | Obese NAFLD:At the family level: Kiloniellaceae/Pasteurellaceae (belonging to Proteobacteria phylum)↑Lactobacillaceae/Lachnospiraceae/Veillonellaceae (belonging to Firmicutes phylum)↑Ruminococcaceae(belonging to Firmicutes phylum)↓ Porphyromonadaceae (belonging to Bacteroidetes phylum)↓At the genus level: Lactobacillu (belonging to Lactobacillaceae family)↑ Robinsoniella/Roseburia/Dorea (from Lachnospiraceae family)↑ Oscillibacter (from Ruminococcaceae family)↓ | [12] |
| Human | 16S rRNA sequencing | Obese patients without NASH (n=25), NASH (n=22), healthy controls (n=16) | Stool | At the Phylum level: Proteobacteria↑At the family level: Enterobacteriaceae↑ | [13] |
| Human | 454 pyrosequencing of 16S rRNA | lean-NAFLD (n=43) and healthy controls (n=83) | Stool | Lean-NAFLD: Bacteroidetes↑ Firmicutes↓ Lachnospiraceae↓ Ruminococcaceae↓ Lactobacillaceae↓ Peptostreptococcaceae↓ Coprococcus↓ Anaerosporobacter↓ Anaerotruncus↓ Ruminococcus↓ Lactobacillus↓ | [14] |
| Human | 16S rRNA sequencing | NASH patients (n=13)Healthy controls (n=10) | Stool | lean NASH: Faecalibacterium↓ Ruminococcus↓ Lactobacilli ↓overweight NASH: Bifidobacterium↓obesity NASH: Lactobacilli↑ | [15] |
| Human | 16S rRNA sequencing | Obese children without NAFLD (n=11), NAFLD (n=13), healthy lean children (n=26) | Stool | NAFLD: Epsilonproteobacteria↑ GammaProteyia↑ Prevotella↑ | [16] |
| Human | 454-Junior Genome Sequencerpyrosequencing of 16S rRNA | Pediatric NAFLD (n=27), NASH (n=26), obesity (n=8), healthy (n=54) | Fecal | Pediatric NAFLD: At the phylum level: Actinobacteri↑ Bacteroidetes↓At the family level: Rikenellaceae↓At the genus level: Oscillospira↓ Ruminococcus↑Blautia↑ Bradyrhizobium↑ Anaerococcus↑ Peptoniphilus↑ Propionibacterium acnes↑ Dorea↑ | [17] |
| Human | 16S rRNA sequencing | NASH (n=35), no NASH (n=22) | Stool | NASH: Bacteroides↑ Prevotella↓Significant fibrosis:Ruminococcus↑ Bacteroides↑ Prevotella↓ | [18] |
| Human | Whole-genome shotgun sequencing of DNA | Mild/moderateNAFLD (n=72), aggravated fibrosis (n=14) | Stool | Mild/moderate NAFLD: Firmicutes↑ Bacteroides vulgatus↑ Eubacterium rectale↑Aggravated fibrosis: Proteobacteria↑ Bacteroides vulgatus↑ Escherichia coli ↑Ruminococcus obeum ↓ Eubacterium rectale↓ | [19] |
From the above studies, we found that there are significant differences in microbiome between healthy people and NAFLD or NASH patients. First, obesity and age may lead to different characteristics of intestinal bacteria among patients with NAFLD. Second, the characteristics of intestinal bacteria are also different in different disease stages of NAFLD. However, the potential mechanisms involved in the role of microbiota in NAFLD are still controversial, it is necessary to clarify the specific mechanisms and the therapeutic effect of the above bacteria on NAFLD in the further study.
1.2Gut bacteria taxa in alcoholic fatty liver diseaseStudies have shown that the consumption of alcohol changes intestinal bacteria [20,21]. For example, acute alcohol intake in Swiss mice increased the total amounts of Enterobacteria compared with control mice, suggesting that alcohol intake caused intestinal dysbacteriosis and liver injury [22]. Lieber-Decarli liquid diet containing alcohol (5% v/v) for 8 weeks in C57BL/6N mice led to an increase in Proteobacteria and Actinobacteria and a decreased level of Bacteroidetes and Firmicutes, and alcohol causes a selective expansion of microbial membership, intestinal permeability and bacterial translocation, increase in plasma endotoxin, fecal pH, hepatic inflammation and injury [23]. Many factors are involved in alcohol consumption, including drinking duration, different types of alcohol, and the severity and degrees of alcohol-induced liver lesions. Here, we will review the gut microbiota composition in the conditions described above.
First, different degrees of alcoholic liver disease are associated with different intestinal bacteria compositions. In a human study, 38 alcoholic patients were enrolled and divided into three groups: no alcoholic-induced liver lesions (noAH), non-severe AH (nsAH), and severe AH (sAH) [24]. Patients with sAH had higher proportions of Bifidobacteria and Streptococci and lesser amounts of Atopobium compared with patients with noAH. In addition, a high proportion of Enterobacteria was observed in patients with AH, while an abundance of Bifidobacteria, Streptococci, Enterobacteria and Atopobium showed no difference between noAH and nsAH by 454 pyrosequencing. This study shows that individual susceptibility to ALD is substantially driven by intestinal microbiota [24]. Another human study showed that a higher proportion of Proteobacteria and decreased amounts of Bacteroidetes and Verrucomicrobia were observed in alcoholics with or without alcoholic liver disease compared to healthy controls using 16S rRNA sequencing, and suggests that the dysbiotic microbiome could contribute to endotoxemia in alcoholics [25]. Furthermore, patients with AH gained improved liver function, including decreased levels of aspartate aminotransferase, bilirubin, prothrombin time and other indicators after eating Lactobacillus subtilis/Streptococcus faecium (1500mg/day) for seven days [26]. In addition, animal studies provided evidence for alcohol-induced intestinal dysbiosis. Gut microbiota were assessed in mice after alcoholic feeding for 5 weeks. There were 23 genera showing different numbers at the genus level between sAH and noAH mice, with a higher abundance of Bacteroides, Bilophila, Alistipes, Butyricimonas and Clostridium cluster in sAH mice, while noAH mice had higher amounts of Barnesiella, Parasutterella and Alphaproteobacteria, sAH mice had higher intestinal permeability and greater translocation of bacteria to mesenteric lymph nodes than noAH mice [24].
Second, the duration of alcohol consumption and drinking doses were related to the composition of intestinal bacteria. In an animal study, alcohol or dextrose was given to male Sprague-Dawley (SD) rats by gavage for 4, 6 or 10 weeks. The study found no differences in microbiota composition from ileum and colon in alcohol-fed rats compared to dextrose-fed rats at week 4 and 6, while significant differences were observed after 10 weeks of alcohol treatment (2g/kg/day for two weeks, 8g/kg/day for 8 weeks) using length heterogeneity PCR (LH-PCR) [27]. Unfortunately, the study did not clarify specific bacteria taxa at different levels. However, another animal study addressed this question. C57/B6 mice were fed with ethanol via gavage for 1 day, 1 week and 3 weeks (final ethanol dose, 30.9g/kg/day), and bacteria contents of the small intestine and large intestine were analyzed by 16S rRNA gene amplicon sequencing [28]. The study demonstrated reduced amounts of Firmicutes, namely, Lactococcus, Pediococcus, Lactobacillus and Leuconostoc genera, and increased proportions of Bacteroidetes and Verrucomicrobia, such as Bacteroidales, Bacteroides and Porphyromonadaceae, in the 10-week-ethanol group, while no difference was observed in the 1 day and 1 week groups. This study also shows that alcohol causes intestinal bacterial overgrowth and enteric dysbiosis [28]. A human study explored the gut bacteria and permeability in alcohol-dependent (AD) subjects and demonstrated that AD subjects with high intestinal permeability had a lower abundance of Ruminococcaceae and higher amounts of Lachnospiraceae compared to AD subjects with low intestinal permeability and controls at the family level, and showed that the gut microbiota could alter the gut-barrier function and influence behavior in alcohol dependence [29]. The imbalance of intestinal microflora in patients with alcoholism is related to the decrease of Bacteroidetes abundance and the increase of Enterobacteriaceae and Proteobacterium abundance [30].
Third, different types of alcohol and different immune characteristics lead to different structures of intestinal microbiota. Ten healthy male adults were enrolled and divided into four consecutive periods, including a washout period without alcohol intake for 15 days and 3 periods of feeding with only de-alcoholized red wine (272ml/d), red wine (272ml/d), or gin (100ml/d) for 20 days [31]. Fetal samples were collected in each period and were analyzed using quantitative real-time PCR. The study detected increased levels of the phylum Proteobacteria, Fusobacteria, Firmicutes, and Bacteroidetes in the red wine intake period compared to the gin consumption period, and they found that red wine polyphenols can inhibit non-beneficial bacteria from the human microbiota and potentiate the growth of probiotic bacteria such as bifidobacteria [31]. At the genus and species levels, increased numbers of the Enterococcus genus, Prevotella genus, Bacteroides genus, Bifidobacterium genus, Eggerthella lenta species, and Blautia coccoides-Eubacterium rectale species and decreased numbers of the Clostridium genus and Clostridium histolyticum species were found in the red wine and de-alcoholized red wine consumption periods compared with the gin-intake period [31]. In an animal experiment, Female C57BL/6J mice were divided into AlcRes(alcohol-resistant mice) group, and AlcSens(alcohol-sensitive mice) group. In AlcSens group, decreased Bacteroidetes and Proteobacteria was found, while Actinomyces and Firmicutes increased, and alcohol-induced liver damage is associated with low levels of Bacteroides. While AlcRes group was dominated with Coprococcus genus of the Firmicutes phylum and the Moraxellaceae and Helicobacteraceae families of the Proteobacteria phylum. Ethanol induced steatosis and liver inflammation, which were associated with disruption of gut homeostasis, in alcohol-sensitive, but not alcohol resistant mice. Pectin treatment and fecal microbiota transplantation prevented steatosis, liver inflammation, and restored gut homeostasis [32].
In conclusion, increased Enterobacteria and Proteobacteria and Bacteroidetes and Actinobacteria were observed in AFLD. The proportion of Firmicutes remains controversial (Fig. 1, Table 2). In addition, individuals have differential responses to alcohol, and the characteristics of intestinal flora are different. Different types of alcohol, drinking time, alcohol consumption have different effects on intestinal flora. Consequently, specific microbiota should be used in specific causes-induced AFLD.
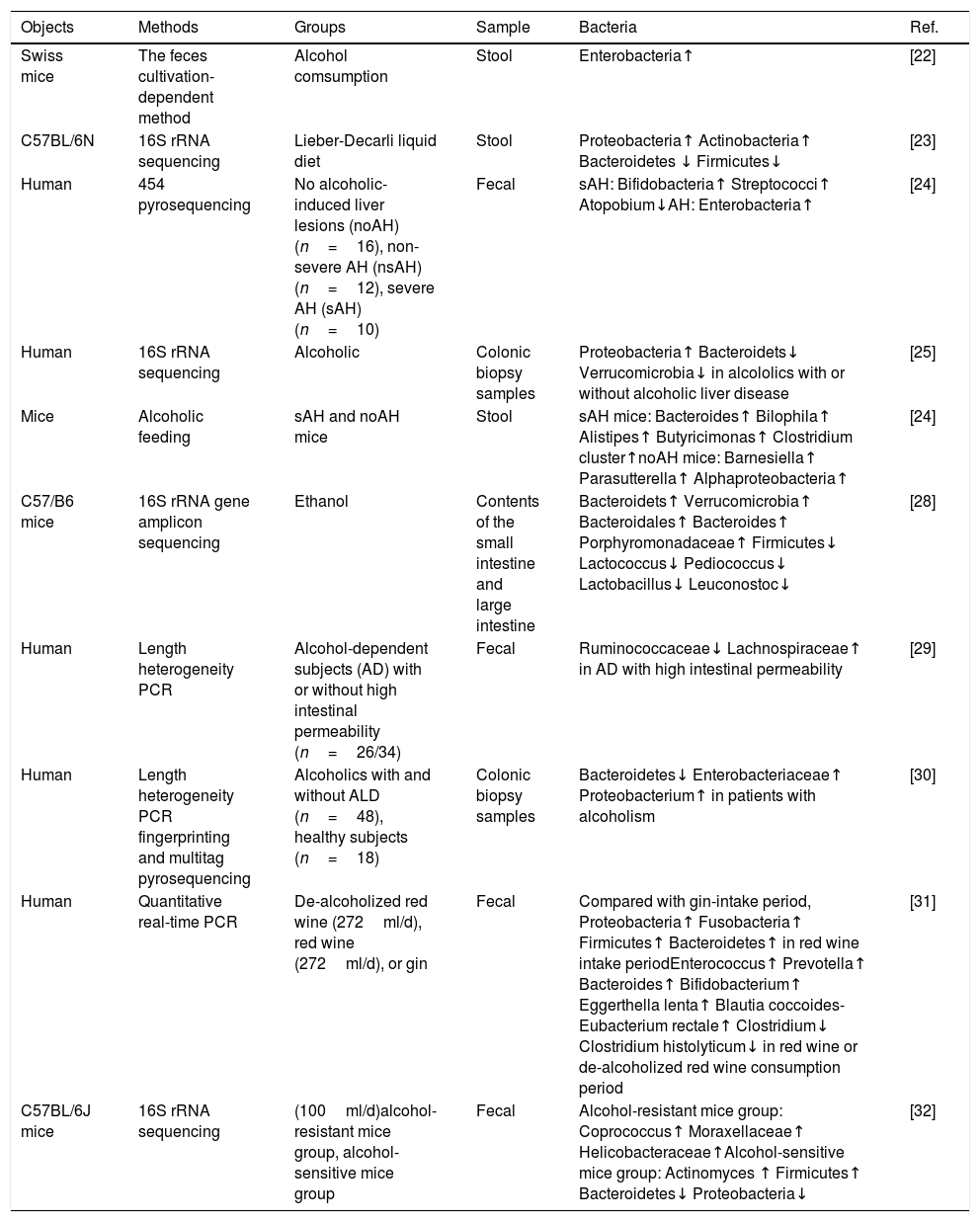
Characteristics of stool bacteria in alcoholic fatty liver disease.
| Objects | Methods | Groups | Sample | Bacteria | Ref. |
|---|---|---|---|---|---|
| Swiss mice | The feces cultivation-dependent method | Alcohol comsumption | Stool | Enterobacteria↑ | [22] |
| C57BL/6N | 16S rRNA sequencing | Lieber-Decarli liquid diet | Stool | Proteobacteria↑ Actinobacteria↑ Bacteroidetes ↓ Firmicutes↓ | [23] |
| Human | 454 pyrosequencing | No alcoholic-induced liver lesions (noAH) (n=16), non-severe AH (nsAH) (n=12), severe AH (sAH) (n=10) | Fecal | sAH: Bifidobacteria↑ Streptococci↑ Atopobium↓AH: Enterobacteria↑ | [24] |
| Human | 16S rRNA sequencing | Alcoholic | Colonic biopsy samples | Proteobacteria↑ Bacteroidets↓ Verrucomicrobia↓ in alcololics with or without alcoholic liver disease | [25] |
| Mice | Alcoholic feeding | sAH and noAH mice | Stool | sAH mice: Bacteroides↑ Bilophila↑ Alistipes↑ Butyricimonas↑ Clostridium cluster↑noAH mice: Barnesiella↑ Parasutterella↑ Alphaproteobacteria↑ | [24] |
| C57/B6 mice | 16S rRNA gene amplicon sequencing | Ethanol | Contents of the small intestine and large intestine | Bacteroidets↑ Verrucomicrobia↑ Bacteroidales↑ Bacteroides↑ Porphyromonadaceae↑ Firmicutes↓ Lactococcus↓ Pediococcus↓ Lactobacillus↓ Leuconostoc↓ | [28] |
| Human | Length heterogeneity PCR | Alcohol-dependent subjects (AD) with or without high intestinal permeability (n=26/34) | Fecal | Ruminococcaceae↓ Lachnospiraceae↑ in AD with high intestinal permeability | [29] |
| Human | Length heterogeneity PCR fingerprinting and multitag pyrosequencing | Alcoholics with and without ALD (n=48), healthy subjects (n=18) | Colonic biopsy samples | Bacteroidetes↓ Enterobacteriaceae↑ Proteobacterium↑ in patients with alcoholism | [30] |
| Human | Quantitative real-time PCR | De-alcoholized red wine (272ml/d), red wine (272ml/d), or gin | Fecal | Compared with gin-intake period, Proteobacteria↑ Fusobacteria↑ Firmicutes↑ Bacteroidetes↑ in red wine intake periodEnterococcus↑ Prevotella↑ Bacteroides↑ Bifidobacterium↑ Eggerthella lenta↑ Blautia coccoides-Eubacterium rectale↑ Clostridium↓ Clostridium histolyticum↓ in red wine or de-alcoholized red wine consumption period | [31] |
| C57BL/6J mice | 16S rRNA sequencing | (100ml/d)alcohol-resistant mice group, alcohol-sensitive mice group | Fecal | Alcohol-resistant mice group: Coprococcus↑ Moraxellaceae↑ Helicobacteraceae↑Alcohol-sensitive mice group: Actinomyces ↑ Firmicutes↑ Bacteroidetes↓ Proteobacteria↓ | [32] |
In a study investigating the relationship between dysbiosis and fibrogenesis, C57 BL/6 mice fed with control (CTRL) or HFD underwent bile duct ligation (BDL) or CCL4intraperitoneal injection [33]. The HFD+BDL group had increased fibrosis, a reduction in Bacteroidetes and increased Firmicutes and Proteobacteria [33].
A human study was performed to study the characteristics of gut microbiota in patients of cirrhosis with HCC; the analysis of stool samples from 15 cirrhosis patients with HCC and 15 without HCC showed that Escherichia coli was over-represented in patients with HCC, while others, such as Enterobacteriaceae and Lactobacillus species, showed no significant differences between the two groups, the study showed that the overgrowth of Escherichia coli may be related to the occurrence of liver cancer [34]. Different results were found in another human study, in which 36 patients with cirrhosis (24 hepatitis B-related, 12 alcohol-related) and 24 healthy controls were selected. The results showed reduced Bacteroidetes and increased Proteobacteria and Fusobacteria in patients with cirrhosis at the phylum level. At the family level, patients with cirrhosis had a higher proportion of Enterobacteriaceae, Veillonellaceae, and Staphylococcae [35]. In addition, Staphylococcae and Lachnospiraceae showed positive and negative correlation with Child-Pugh score, respectively [35]. Similar results were found in another clinical study. Stool bacteria analysis from five alcohol drinkers and five non-alcoholic cirrhotics revealed an increase in Veillonellaceae from phylum Firmicutes and decrease in Bacteroidaceae and Porphyromonadaceae from Bacteroidetes in drinkers [36]. Another study used shotgun metagenomics to analyzed gut microbiota from patients with alcoholic cirrhosis and healthy controls, and showed the abundance of Parabacteroides, Prevotella, Clostridium and Bacteroides decreased and the abundance of Lactobacillus and Bifidobacteria increased in patients with alcoholic cirrhosis, this study suggests that the hallmarks of the liver cirrhosis are likely linked to the impaired bile secretion [37]. Another study of alcoholic cirrhotic liver showed a higher abundance of Enterobacteriaceae and Halomonadaceae and lower amounts of Lachnospiraceae, Ruminococcaceae and Clostridiales XIV in alcoholic cirrhotics compared with those without alcoholic etiology using multi-tagged pyrosequencing [38]. Different from patients with alcoholic-related cirrhosis, NASH patients had higher Porphyromonadaceae and Bacteroidaceae and lower Veillonellaceae [38]. Furthermore, the relationship between MELD score and bacteria was also analyzed, suggesting that Clostridiales XIV, Lachnospiraceae, Ruminococcaceae, and Rikenellaceae negatively correlated with MELD score, while Staphylococcae, Enterococcaceae and Enterobacteriaceae positively correlated with MELD score [38]. Another human study showed that different gut microbiota were observed in compensated/decompensated patients with cirrhosis and controls from Turkey and the United States using 16S rRNA sequencing [39]. In the groups within the Turkish cohort, patients with decompensated cirrhosis had higher contents of Streptococcaceae and Sutterellaceae, and lower contents of Enterococcaceae, compared with compensated cirrhosis group; Patients with compensated or decompensated cirrhosis had higher Enterococcaceae and Streptococcaceae compared with controls [39]. In the US cohort, the decompensated cirrhosis patients had higher Lactobacillaceae, Veillonellaceae, and Fusobacteria compared with controls group; and compensated cirrhosis patients had lower levels of Veillonellaceae and Bifidobacteriaceae and higher autochthonous taxa compared with decompensated cirrhosis patients group [39]. Furthermore, Turkish cirrhosis patients owed higher levels of beneficial taxa such as Ruminococcaceae and other Clostridiales and Bifidobacteriaceae, and lower levels of Enterococcaceae when compared with cirrhosis patients from the United States. They concluded that diet was strongly associated with microbial composition in cirrhosis, and the plant phenol-containing dietary compounds, such as tea, coffee, and chocolate, had beneficial effects on microbial diversity[39]. Similarly, another human study showed that gut microbiota were observed in healthy controls (n=47), compensated/decompensated liver cirrhosis patients (n=49/46) using contained paired-end metagenomic sequencing. Compared to healthy controls, at the genera level, Alistipes, Odoribacter, Eubacterium, Ruminococcus, Eubacterium rectale, Alistipes putredinis, Alistipes shahii, Coprococcus eutactus, Roseburia intestinalis, Clostridiumsp and Bacteroides intestinalis were significantly less abundant in compensated/decompensated patients, while Veillonella, Streptococcus, Lactobacillus, Megasphaera, Haemophilus, Haemophilus parainfuenzae, Streptococcus salivarius, Lactobacillus salivarius, Veillonella parvula, Ruminococcus gnavus and Bifidobacterium dentium were more abundant. It also showed that the further downregulation of Tannerella, Bilophila, Alistipes indistinctus, Bilophila wadsworthia, Bilophila, Ruminococcus champanellensis, Tannerella, Clostridium botulinum, Clostridium leptum, Clostridium methylpentosum, Clostridium and upregulation of Veillonella, Veillonella atypica, Veillonella, Veillonella dispar, and Veillonella sp. oral taxon 158 continued during disease progression from compensation to decompensation stage [40].
In conclusion, the increase of Proteobacteria and the decrease of Bacteroidetes were observed in liver cirrhosis. The proportion of Firmicutes remains controversial (Fig. 1, Table 3).
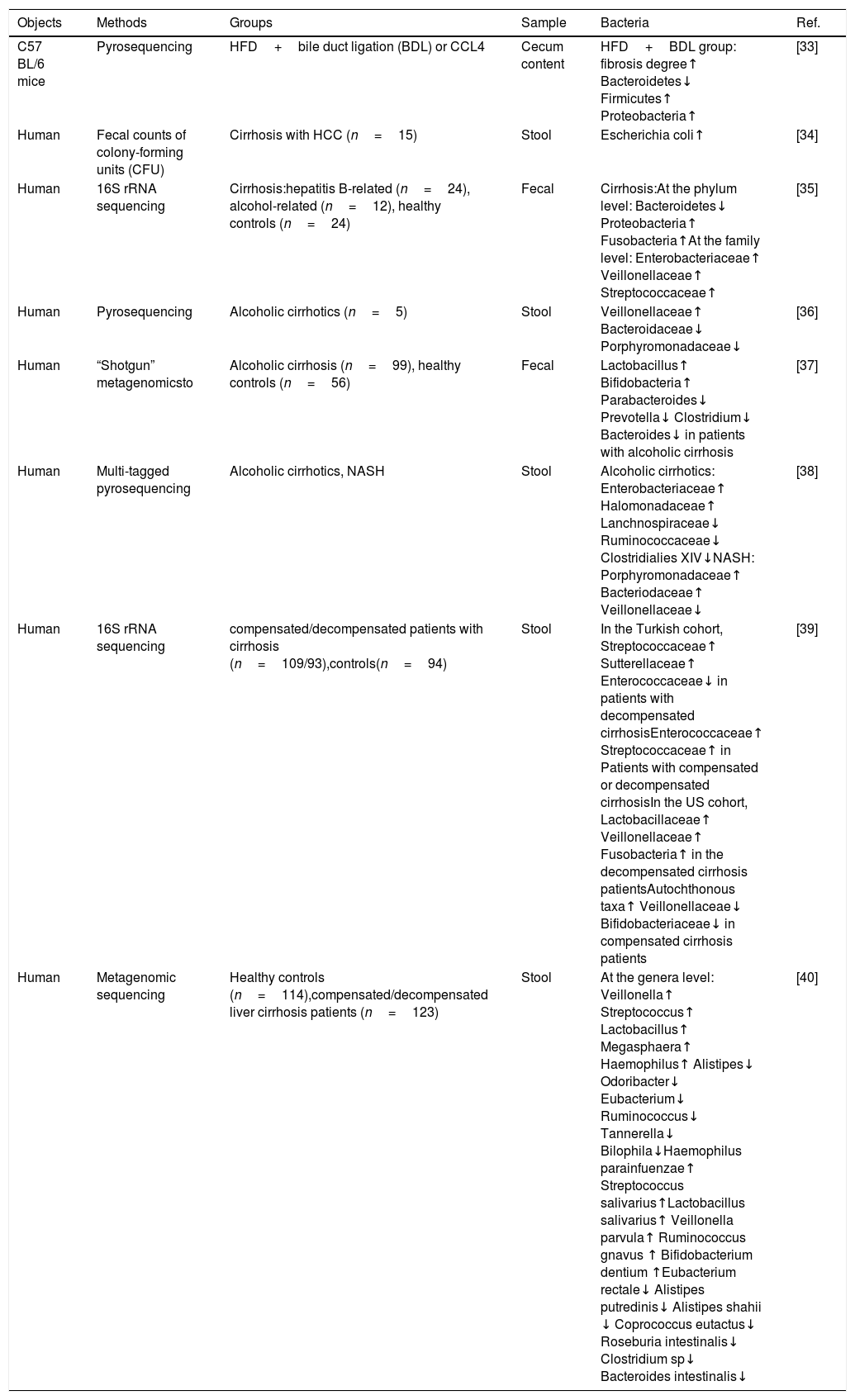
Characteristics of stool bacteria in Liver cirrhosis.
| Objects | Methods | Groups | Sample | Bacteria | Ref. |
|---|---|---|---|---|---|
| C57 BL/6 mice | Pyrosequencing | HFD+bile duct ligation (BDL) or CCL4 | Cecum content | HFD+BDL group: fibrosis degree↑ Bacteroidetes↓ Firmicutes↑ Proteobacteria↑ | [33] |
| Human | Fecal counts of colony-forming units (CFU) | Cirrhosis with HCC (n=15) | Stool | Escherichia coli↑ | [34] |
| Human | 16S rRNA sequencing | Cirrhosis:hepatitis B-related (n=24), alcohol-related (n=12), healthy controls (n=24) | Fecal | Cirrhosis:At the phylum level: Bacteroidetes↓ Proteobacteria↑ Fusobacteria↑At the family level: Enterobacteriaceae↑ Veillonellaceae↑ Streptococcaceae↑ | [35] |
| Human | Pyrosequencing | Alcoholic cirrhotics (n=5) | Stool | Veillonellaceae↑ Bacteroidaceae↓ Porphyromonadaceae↓ | [36] |
| Human | “Shotgun” metagenomicsto | Alcoholic cirrhosis (n=99), healthy controls (n=56) | Fecal | Lactobacillus↑ Bifidobacteria↑ Parabacteroides↓ Prevotella↓ Clostridium↓ Bacteroides↓ in patients with alcoholic cirrhosis | [37] |
| Human | Multi-tagged pyrosequencing | Alcoholic cirrhotics, NASH | Stool | Alcoholic cirrhotics: Enterobacteriaceae↑ Halomonadaceae↑ Lanchnospiraceae↓ Ruminococcaceae↓ Clostridialies XIV↓NASH: Porphyromonadaceae↑ Bacteriodaceae↑ Veillonellaceae↓ | [38] |
| Human | 16S rRNA sequencing | compensated/decompensated patients with cirrhosis (n=109/93),controls(n=94) | Stool | In the Turkish cohort, Streptococcaceae↑ Sutterellaceae↑ Enterococcaceae↓ in patients with decompensated cirrhosisEnterococcaceae↑ Streptococcaceae↑ in Patients with compensated or decompensated cirrhosisIn the US cohort, Lactobacillaceae↑ Veillonellaceae↑ Fusobacteria↑ in the decompensated cirrhosis patientsAutochthonous taxa↑ Veillonellaceae↓ Bifidobacteriaceae↓ in compensated cirrhosis patients | [39] |
| Human | Metagenomic sequencing | Healthy controls (n=114),compensated/decompensated liver cirrhosis patients (n=123) | Stool | At the genera level: Veillonella↑ Streptococcus↑ Lactobacillus↑ Megasphaera↑ Haemophilus↑ Alistipes↓ Odoribacter↓ Eubacterium↓ Ruminococcus↓ Tannerella↓ Bilophila↓Haemophilus parainfuenzae↑ Streptococcus salivarius↑Lactobacillus salivarius↑ Veillonella parvula↑ Ruminococcus gnavus ↑ Bifidobacterium dentium ↑Eubacterium rectale↓ Alistipes putredinis↓ Alistipes shahii ↓ Coprococcus eutactus↓ Roseburia intestinalis↓ Clostridium sp↓ Bacteroides intestinalis↓ | [40] |
From the above mentioned results, we found various studies showing different bacteria taxa leading to cirrhosis. First, various methods for bacteria analysis might lead to different results. Second, various factors related to cirrhosis might also participate. Third, different cirrhosis stages might be associated with different bacterial characteristics. Therefore, further studies regarding the role of these bacteria in the prevention or treatment of cirrhosis should be performed.
2ConclusionsIncreased amounts of Lachnospiraceae from the phylum Firmicutes and Roseburia from the Lachnospiraceae family were associated with NAFLD, and alcohol intake increased the proportion of Enterobacteria and Proteobacteria. Reduced Bacteroidetes was observed in cirrhosis. Microbiota can improve or aggravate three kinds of liver diseases through several mechanisms, including glucose homeostasis disorder, increased liver lipid metabolism, increased alcohol production, increased energy metabolism, increased intestinal permeability, bacterial translocation, increased plasma endotoxin, increased fecal pH, intestinal bacterial overgrowth, enteric dysbiosis, gut-barrier function destruction, impaired bile secretion. However, the depletion of these bacteria is needed to verify their role in liver disease.
AbbreviationsNAFLD non-alcoholic fatty liver disease alcoholic fatty liver disease hepatocellular carcinoma
This work was supported by the Youth Foundation of the Affiliated Hospital of Qingdao University [grant number 3091]. Funding sources are not involved in the collection, analysis and interpretation of data, the writing of reports, and the determination to submit articles for publication.
Conflict of interestThe authors have no conflicts of interest to declare.
We thank American Journal Experts for langue polishing service.



