



Treatment of experimental animals with prototypical enzyme inducers represents a useful tool to characterize the role of different isozymes in drug metabolism and to improve our knowledge on factors regulating their synthesis at the transcriptional level. The effect of model enzyme inducers on phase II (conjugating) enzyme families, including UDP-glucuronosyltransferase’s and glutathione-S-transferase’s, has been well characterized in rodent liver. More recently, the effect of inducers on the expression of canalicular multidrug resistance-associated protein 2 (Mrp2) has been focused upon. The identification of a number of conjugated drugs as Mrp2 substrates suggests that both the conjugation and transport systems act coordinately to improve drug elimination from the body. We provide evidence about circumstances resulting in the simultaneous upregulation of phase II enzymes and Mrp2 in hepatic and extrahepatic tissues, most likely involving activation of common nuclear receptors (e.g. FXR, PXR). Additionally, we provide an analysis of examples of drug-induced toxicity leading to the simultaneous downregulation of both systems. Potential therapeutic strategies based on the modulation of expression of these systems are also briefly commented upon.
Abbreviations
Antioxidant/electrophile response element: ARE/ EpRE
Aryl hydrocarbon receptor: AhR
Bile salt export pump: Bsep
Constitutive androstane receptor: CAR
Glutathione-S-transferase: GST
Lipopolysaccharide: LPS
Multidrug resistance-associated protein 2: Mrp2
Peroxisome proliferator-activated receptor: PPAR
Phenobarbital: PB
Pregnane X receptor: PXR
Spironolactone: SL
UDP-glucuronosyltransferase: UGT
Ursodeoxycholic acid: UDC
9-cis-retinoic acid receptor: RXR/RAR
Financial support: This study was supported by CONICET, ANPCYT, UNR and Fundación Antorchas, Argentina.
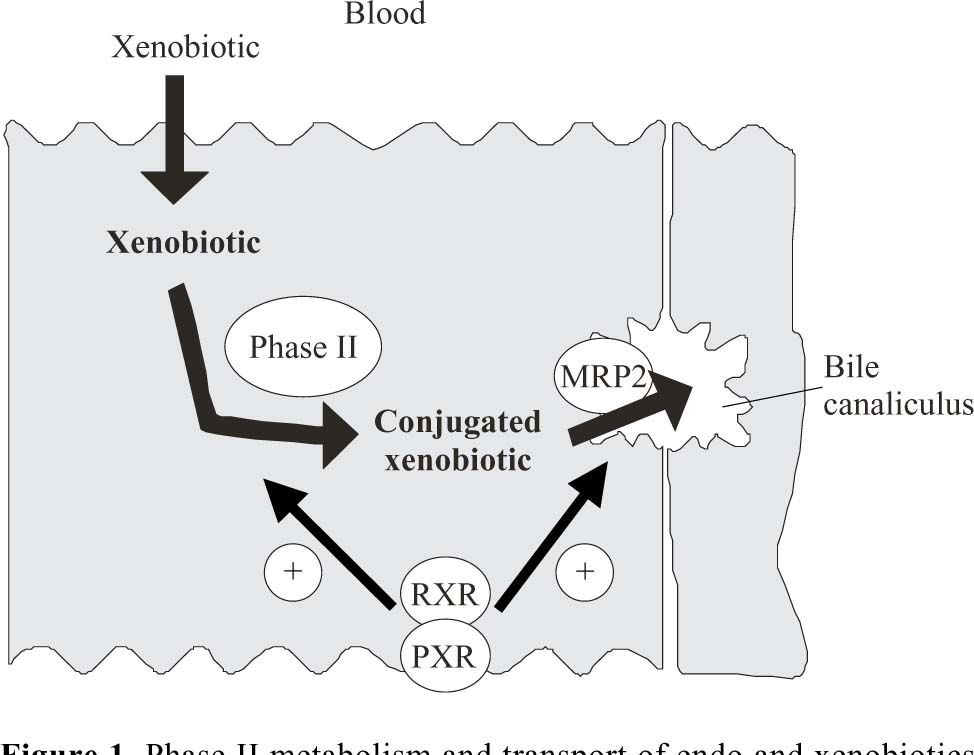
IntroductionThe phase II or post-oxidative biotransformation system comprises an array of enzymes that catalyze the incorporation of a new hydrophilic endogenous group into hydrophobic molecules, thereby decreasing its polarity and eventual toxicity. Phase II reactions can be catalyzed by a number of different enzyme families including UDP-glucuronosyltransferase (UGT), glutathione S-transferase (GST), sulphotransferase and methytransferase. The multidrug resistant proteins (MRPs) represent a family of ATP-dependent transporters that are expressed normally in epithelial cells, but which exhibit elevated expression levels in tumoral cells. Mrp2, one member of the MRP family, is preferentially localized in the apical membrane of hepatocytes, 1 renal tubular cells2 and enterocytes.3 Mrp2 is involved in the secretion of a large number of conjugated compounds, with most Mrp2 substrates being conjugated derivatives of endogenous compounds, drugs and carcinogens (Figure 1).1 Consequently, they may act coordinately to eliminate these compounds from the body.
 or in the cytosol by Glutathione-S-Transferase (GST) or sulfotransferase, the product may be secreted into bile by Mrp2. The nuclear Pregnane X Receptor (PXR) is activated by a large number of endogenous and exogenous chemicals including steroids, antibiotics, antimycotics, bile salts, etc, thus serving as a generalized sensor of hydrophobic toxins. Generally, the transcriptional activation via CAR and PXR involves the formation of heterodimers between these receptors and 9-cis-retinoic acid receptor (RXR). Then, the heterodimer bind to response elements in the regulatory regions of target genes.")
Phase II metabolism and transport of endo and xenobiotics in the liver and their regulation by nuclear receptors. Hydrophobic xenobiotics may enter the cell by diffusion through the basolateral membrane in the hepatocyte. After conjugation in the endoplasmic reticulum by UDP-glucuronosyltransferase (UGT) or in the cytosol by Glutathione-S-Transferase (GST) or sulfotransferase, the product may be secreted into bile by Mrp2. The nuclear Pregnane X Receptor (PXR) is activated by a large number of endogenous and exogenous chemicals including steroids, antibiotics, antimycotics, bile salts, etc, thus serving as a generalized sensor of hydrophobic toxins. Generally, the transcriptional activation via CAR and PXR involves the formation of heterodimers between these receptors and 9-cis-retinoic acid receptor (RXR). Then, the heterodimer bind to response elements in the regulatory regions of target genes.
On any given day, an average person may be exposed to literally thousands of different molecules. Some of these substances, such as carbohydrates and proteins, are necessary sources of nutrition and are processed in refined biochemical systems. However, humans and other biological organisms are also exposed throughout their lifetime to a vast array of xenobiotics including pharmacological drugs, environmental pollutants and natural toxins. The major routes of exposure to foreign chemicals include ingestion in the diet, inhalation or absorption (e.g. skin, GI). More specifically, oral exposure can occur via the ingestion of food and drugs or by swallowing part of the inhaled xenobiotics after clearance from the tracheobronchial tree.4 Most xenobiotics to some degree undergo a first-pass metabolism in which their biotransformation within enterocytes and hepatocytes prevents their distribution throughout the body, and thereby facilitates their clearance. Hepatic first-pass clearance has been extensively studied,5 in addition, the role of the small intestine as an intrinsic component of presystemic elimination of xenobiotics has also been elucidated.6 Chemicals absorbed into the portal system are transported to the liver, the major site of biotransformation in the body. Nifedipine for example, a well-absorbed drug, is subjected to first-pass phase I metabolism in humans resulting in an oral bioavailability of less than 50%.7 Similarly, phase II reactions in liver and intestine (mediated by UGT and GST isoenzymes) also play a significant role in first-pass clearance of drugs and dietary xenobiotics.8-11 For example, following oral administration, morphine undergoes significant first-pass metabolism through conjugation with glucuronic acid thus leading to the 3-and 6-derivatives, in addition to oxidative metabolism.12 Hepatic and intestinal production of morphine-6-glucuronide is particularly relevant because of the high rate of synthesis and activity that is known to be greater than the parent compound. 13 Numerous other drugs including ethynylestradiol (EE), salicilic acid, paracetamol, chloramphenicol, propanolol and oxacepan, also undergo first-pass conjugation reactions, and are predominantly catalyzed by members of the UGT enzyme family in both liver and intestine. 14-16 Following phase II metabolism, the conjugated derivatives are secreted through the apical membrane in a process mediated by Mrp2.
Normal localization and function of Phase II enzymes and Mrp2Liver cells express both phase II enzymes and Mrp2, thereby accounting for the metabolism and excretion of a wide variety of endogenous and exogenous compounds in the hepatocyte. In many instances, conjugation is a prerequisite for the compound to be recognized by the transporter. For example, bilirubin requires conjugation with glucuronic acid before it can be secreted into bile.17 The biotransformation of compounds within the liver generally culminates in a variety of oxidative (e.g. hydroxylated), conjugated or mixed substrates. Recently, Dietrich et al reported that Mrp2 is involved in the secretion into bile of several different metabolites of 2-amino-1-methyl-6-phenyl-imidazo[4,5-b]pyridine (PhIP), the most abundant carcinogen in overcooked meat.18 The authors compared the biliary excretion of intact and hydroxylated PhIP glucuronides, the main derivatives of PhIP, between normal and Mrp2 genetically-deficient (TR-) rats, and observed that their secretion was decreased in TR-animals by almost 50%. The substrates exhibiting the highest Vmax/ Km values for Mrp2, among a range of compounds tested, were the glutathione derivatives of leukotriene C4 (LTC4), dinitrophenyl-glutathione (GS-DNP), estradiol 17β-glucuronide and bilirubin mono-and di-glucuronides. 1 Therefore, conjugated derivatives are not the unique, but probably the preferential substrates for Mrp2.
Immunohistochemical analyses in the normal rat liver have demonstrated that UGTs are localized in both periportal and perivenous hepatocytes, and that Mrp2 is also expressed in both regions.19,20 Studies on the localization of UGTs and MRPs in the kidney have also suggested that they may work in cooperation. Several different UGT and Mrp2 isoforms have also been localized to tubular cells from the renal cortex in rats.20,21 A similar distribution profile was observed in the human kidney.22,23 In the rat intestine, glucuronic acid, glutathione and sulfate conjugation of endogenous and exogenous substrates represents the highest activities in the proximal portion with a decrease observed further down the intestinal tract.24-26 Furthermore, a gradient in UGT and GST enzyme activity has been demonstrated along the villus-crypt axis with the highest activities at the tip of the villus and the lowest at the crypt region.20,27 A similar distribution along the small intestine and villus-crypt axis has been reported for intestinal Mrp2.3
The evidence clearly indicates the overlapping localization and coordinated function between phase II enzymes and the apical export pump Mrp2, and thereby facilitating the elimination of potentially harmful endogenous substrates and xenobiotics from the body.
Expression and activity of phase II enzymes and Mrp2 in response to inducers. Role of nuclear receptorsThe expression and activity of enzymatic systems and transporters can be affected by many factors, including enzyme inducers, aging, diet, disease and hormones.10,15,28-30 Regulation may occur either at the transcriptional level, resulting in changes in mRNA and protein contents, or at the level of post-translational processing. In general, the major mechanism by which inducers increase the rate of biotransformation is by activation through a number of ligand-activated transcription factors. Examples of ligand-activated transcription factors include the aryl hydrocarbon receptor (AhR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), peroxisome pro-liferator-activated receptor (PPAR), 9-cis-retinoic acid receptor (RXR/RAR) and antioxidant/electrophile response element (ARE/EpRE), among others.31-33
It has been well documented that the activity of the conjugating enzymes, UGT and GST, are significantly increased in liver and small intestine following treatment with various chemicals. Generally, these inductions are due to an increased expression of specific isoforms. Phenobarbital (PB) and spironolactone (SL), two prescription drugs, have demonstrated inducing properties on Phase I and II systems as well as on genes encoding transporter proteins. Importantly, the nuclear receptor, CAR, has recently been implicated in mediating responses associated with PB exposure.34 In addition, PXR is activated by a variety of natural steroids and xenobiotics, including pregnenolone-16α-carbonitrile, dexamethasone and SL.35
It has previously been reported that following treatment of animals with PB the protein content, activity and mRNA levels of hepatic and intestinal GST isoforms belonging to the alpha and mu classes were significantly increased. 33,36,37 PB administration was also able to increase the hepatic glucuronidation rate towards different substrates, 38,39 as well as protein content and mRNA levels of specific UGT isoenzymes.40-43
The reported literature on the effect of PB on hepatic Mrp2 expression is contradictory and is dependent upon the experimental model used and the time course studied. For example, Klaassen and Johnson reported that pregnenolone-16α-carbonitrile and PB induced Mrp2 expression in liver, as well as the subsequent biliary excretion of Mrp2 substrates.44 Both compounds were also reported to induce phase II enzyme expression including specific UGT isoforms.45 Furthermore, Kast et al. described that Mrp2 mRNA levels were significantly increased by PB in vitro46 while Patel et al.47 showed that Mrp2 protein expression and functional capacity was decreased 48 h after PB treatment. Finally, Hagenbuch et al.48 reported a modest increase in Mrp2 expression at the RNA level after PB treatment without washout.
Several studies have demonstrated that activation of PXR enhances the expression of hepatic GST and UGT isoenzymes.33,49,50 Studies from our laboratory have showed that GST activity towards 1-chloro-2,4-dinitrobenzene (CDNB), as well as the glucuronidation rates of bilirubin and ethynylestradiol in rat, are increased by SL.51-54 More specifically, western blot analyses revealed increased UGT1A1 (bilirubin), UGT1A5 (3-OH ethynylestradiol), and 2B1 (17β-OH ethynylestradiol) protein levels. Furthermore, a northern blot analysis revealed that the up-regulation observed was at the transcriptional level. A report55 recently showed that activation of PXR and CAR induced hepatic UGT1A1 and UGT1A6 expression, and subsequently increased bilirubin clearance.
To assess whether Mrp2 followed a similar induction profile to phase II enzymes in SL-treated rats, we examined its expression and activity in liver and intestine.
Through western blot analyses we observed that both hepatic and intestinal Mrp2 protein increased significantly in SL-treated rats with respect to controls. An in situ immunofluorescence analysis of Mrp2 expression confirmed the immunoblotting data. Furthermore, an analysis of transport activity in liver revealed that biliary secretion of glutathione, a substrate for Mrp2, was also increased by SL, in agreement with the increased expression of the transporter,56 [unpublished results]. Additional authors have also reported the induction of Mrp2 expression by PXR ligands.35,46,55
Thus, the common localization of phase II enzymes with their transport systems in conjunction with their induction by the same compounds, suggests a coordinate action. A study by Bock et al has provided additional evidence in support of the hypothesis of co-regulation. They reported that treatment of Caco-2 cells with the polyphenolic antioxidants quercetin and t-butylhydroquinone increased Mrp2 expression.58 Selected UGT isoforms involved in the conjugation of phenolic compounds (i.e., UGT1A6) were also induced, leading the authors to postulate that coordinate induction of UGT and Mrp2 may facilitate chemoprotection against phenolic toxins and excretion of conjugates in the intestinal lumen. Finally, Huang et al.59 showed that activation of CAR increases hepatic expression of genes known to be involved in bilirubin metabolism and transport across the apical liver membrane (i.e. GSTA1, UGT1A1 and Mrp2).
Co-regulation of Mrp2 and phase II metabolism in cholestasisCholestasis is a pathological condition where a co-regulation of Mrp2 and Phase II detoxification is observed. Trauner et al. demonstrated that in both extrahepatic and intrahepatic models of cholestasis, Mrp2 is down regulated. 60 Bile duct ligation produced a significant decrease in Mrp2 protein as determined by western blotting 3 days after surgery (47%) and is still present after 7 days (73%). This was correlated with an observed decrease in Mrp2 steady-state mRNA levels. Lipopolysaccharide (LPS)- treated rats, a model of intrahepatic cholestasis in sepsis, exhibited similar results. Specifically, 16 hs. after a single-dose administration of the endotoxin (4 mg/kg bw), Mrp2 protein is reduced by 70% and observed mRNA levels are only 10% of control values. At this time, Mrp2 function is maximally decreased.61-63 The administration of ethynylestradiol to experimental animals represents an additional model of intrahepatic cholestasis. After a 5-day administration of 5 mg/kg bw, Mrp2 function is clearly altered.64-67 More specifically, Mrp2 protein is reduced by 57% but steady-state mRNA levels are not modified by the estrogen.60 It is therefore apparent that different mechanisms are involved in Mrp2 regulation in cholestasis. In summary, LPS and bile duct ligation seem to regulate Mrp2 at the mRNA level, whereas the effect of ethynylestradiol is limited to posttranscriptional events.
Decreased Mrp2 expression and function during cholestasis may have important implications for the excretion of various, potentially toxic endo-and xenobiotics. 1,68 The compounds generally undergo phase II metabolism via GST or UGT enzymes, with the related metabolic processes also being affected in the different models of cholestasis. Bile duct ligation leads to a 33% decrease in the glucuronidation of bilirubin within 48 hs, reaching 70% 8 days after surgery.69 Furthermore, GST enzyme activity measured using CDNB is decreased by 40% 14 days post ligation.70 Endotoxin administration also affects GST and UGT activity.71,72 The mRNAs of GSTA2, A3, M1 and M2 decrease reaching a minimum between 6 to 12 hs. after the administration of LPS (1 mg/kg bw).71
In ethynylestradiol-treated rats, UGT and GST enzyme activities are markedly reduced.61,73 Glucuronidation of ethynylestradiol is significantly decreased, correlating with a decrease in UGT2B1 protein as measured by western blotting.73 Since ethynylestradiol 17β-glucuronide is the postulated mediator of cholestasis,74 this decrease can have a protective aim.
A common mechanism for the down-regulation of phase II enzymes and Mrp2 is suggested by the evidence. In the endotoxin model, and probably in bile-duct ligated rats, inflammatory cytokines may be involved.75 IL-1β participates in Mrp2 mRNA reduction induced by LPS since anakinra, a specific antibody against IL-1β prevents its effect. The mechanism seems to be independent of
RXR/RAR.75 The same cytokine modulates GST at a posttranscriptional level.76 In the estrogen-induced cholestasis model the mechanism of regulation governing phase II enzymes is as yet unknown.
The down-regulation of Mrp2 occurring in cholestasis results in the accumulation of different conjugated species within the hepatocyte. A portion of these metabolites is transported to the plasma via multidrug resistance-associated protein 3 (Mrp3), which is induced during cholestasis.77 A coordinated decrease in enzyme activities tends to moderate the accumulation of conjugates. The disadvantage of the potential increase in non-conjugated species concentration is counterbalanced by a lesser accumulation of conjugates with toxic effects (e.g. estradiol 17β-glucuronide).64,74
Potential therapeutic approaches based upon induction of Phase II enzymes and Mrp2It is well established that certain xenobiotics, including several of the compounds mentioned earlier, induce the expression of specific Phase I and II metabolizing enzymes. Some of these agents have therapeutic applications, though usually at lower doses than those demonstrated to induce metabolizing enzymes in experimental animals. However, for some specific drugs, induction was demonstrated in humans. For example SL, administered as a diuretic to patients with ascitis, increases the depuration of antipyrine, a phenomenon associated with increased activity of specific CYP isoforms.78 As described earlier, several xenobiotics also induce the expression of apical transporters, including Mrp2. One of these agents is rifampicin. It was recently reported that rifampicin induced Mrp2 mRNA and protein levels in human duodenum. 79 Thus, increased elimination of Mrp2 substrates (eg, drug conjugates of other therapeutic agents) into the lumen of the gastrointestinal tract during treatment with rifampicin may represent a new mechanism of drug interactions. Several other studies were specifically focused on the effect of model inducers on transporter proteins. It would be of interest to test the potential of such inducers in counteracting the downregulation of these same proteins occurring in experimental liver diseases (e.g. following estrogen or LPS administration).
An additional therapeutic tool useful for the treatment of liver disease is the administration of hydrophilic bile salts such as ursodeoxycholic acid (UDC), or its taurine derivative, TUDC. Long-term (7-day) feeding of normal mice with UDC induces the upregulation of canalicular Mrp2.80 Furthermore, intraperitoneal coadministration of UDC with the lipophilic, toxic bile salt deoxycholate, counteracted the down-regulation of the canalicular bile salt transporter, Bsep.81 Similar protective effects were observed with UDC on cholic acid-induced decreases in the expression of basolateral transporters, particularly organic anion transporters 1 and 4.82
ConclusionMetabolic biotransformation of endogenous and exogenous compounds renders lipophilic molecules more soluble in water, allowing their excretion in bile, urine or feces. It is likely that under specific circumstances, biotransformation systems and membrane transporters may act in cooperation to increase liver and intestinal first-pass metabolism and prevent toxicity of endogenous compounds or xenobiotics incorporated into the body. Altered rates of metabolism can affect their systemic availability and residence time, and hence its toxicity or therapeutic effect. In the case of therapeutic agents, this can result in a number of undesirable drug-drug interactions. The better understanding of simultaneous regulation of metabolism and transport systems may help to prevent these undesirable effects.
AcknowledgementsThe authors thank Professor Mary Vore for having provided laboratory facilities and guidance to carry out some studies reviewed in this article.



