





Sitosterolemia is an autosomal recessive metabolic disease caused by mutations in ABCG5 or ABCG8 genes which encode for the (ATP)-binding cassette (ABC) transporters that are responsible for the trafficking of xenosterols. Liver involvement is not a recognized manifestation of this disease, and cirrhosis has been reported only once in the medical literature. We describe a fatal case of a 21-year old South Asian male who presented with decompensated cirrhosis, and biochemical abnormalities consistent with sitostero-lemia. Genetic testing showed a homozygous pathogenic mutation in ABCG5, confirming the diagnosis. Sitosterolemia is a rare, but likely under-recognized condition, and a high degree of suspicion is imperative to make the diagnosis. We propose that sitosterolemia should be included in the differential diagnosis for patients with cryptogenic cirrhosis, especially as there are effective oral therapies to treat this condition. Newly diagnosed sitosterolemia patients should undergo a thorough hepatology evaluation and follow-up to evaluate for the presence, development, and progression of any hepatic involvement.
Sitosterolemia is a rarely reported disorder of lipid metabolism, first described in 1974,1 in which plant sterols accumulate in blood and tissues due to a defect in their intestinal and hepatic excretion, resulting in variable clinical manifestations.2 Physiologically, small amounts of ingested phytosterols are absorbed and circulated in the plasma,3 but are rapidly cleared by the liver. Plant sterols comprise the major dietary xenosterols humans are exposed to. In this autosomal recessive disease, these absorbed xenoster-ols are not cleared, but accumulate leading to tissue damage. Patel, et al.4 mapped the disease locus, STSL, to chromosome 2p21. Two genes, ABCG5 and ABCG8, comprise the STSL locus, encoding adenosine triphos-phate (ATP)-binding cassette (ABC) transporters stero-lin-1 and sterolin-2, respectively, and biallelic pathogenic variations in ABCG5 or ABCG8 lead to increased intestinal absorption of phytosterols.5-7
Interestingly, it is estimated that less than 100 individuals with sitosterolemia have been described to date, and only one case of cirrhosis reported since the discovery of this disease.8 We describe the case of a 21-year old male who presented with decompensated cirrhosis, and profound hemolytic anemia. Given that he had some classic associations of sitosterolemia, including hemolytic anemia, xanthomas, and ischemic heart disease, we examined his plasma sterol profile, and the biochemical diagnosis of sitosterolemia was made.8 Gene sequencing of ABCG5 and ABCG8 confirmed the diagnosis, revealing a ho-mozygous pathogenic variation in ABCG5.
Case DescriptionA 21-year old male of Pakistani decent presented to an outside hospital with jaundice, chest pain, dyspnea on exertion, as well as worsening ascites and was referred for management of cirrhosis and liver transplantation evaluation. His Model for End-Stage Liver Disease (MELD) score was 32, and was given a presumptive diagnosis of end-stage autoimmune hepatitis. A coronary CT angiogra-phy showed diffuse severe coronary disease without significant calcium deposition, leading the providers to a diagnosis of vasculitis. He was therefore treated with high dose corticosteroids for 8 weeks, without improvement on repeat coronary evaluation. Liver transplant was denied considering the lack of a unifying diagnosis for his combined ischemic heart disease and liver failure.
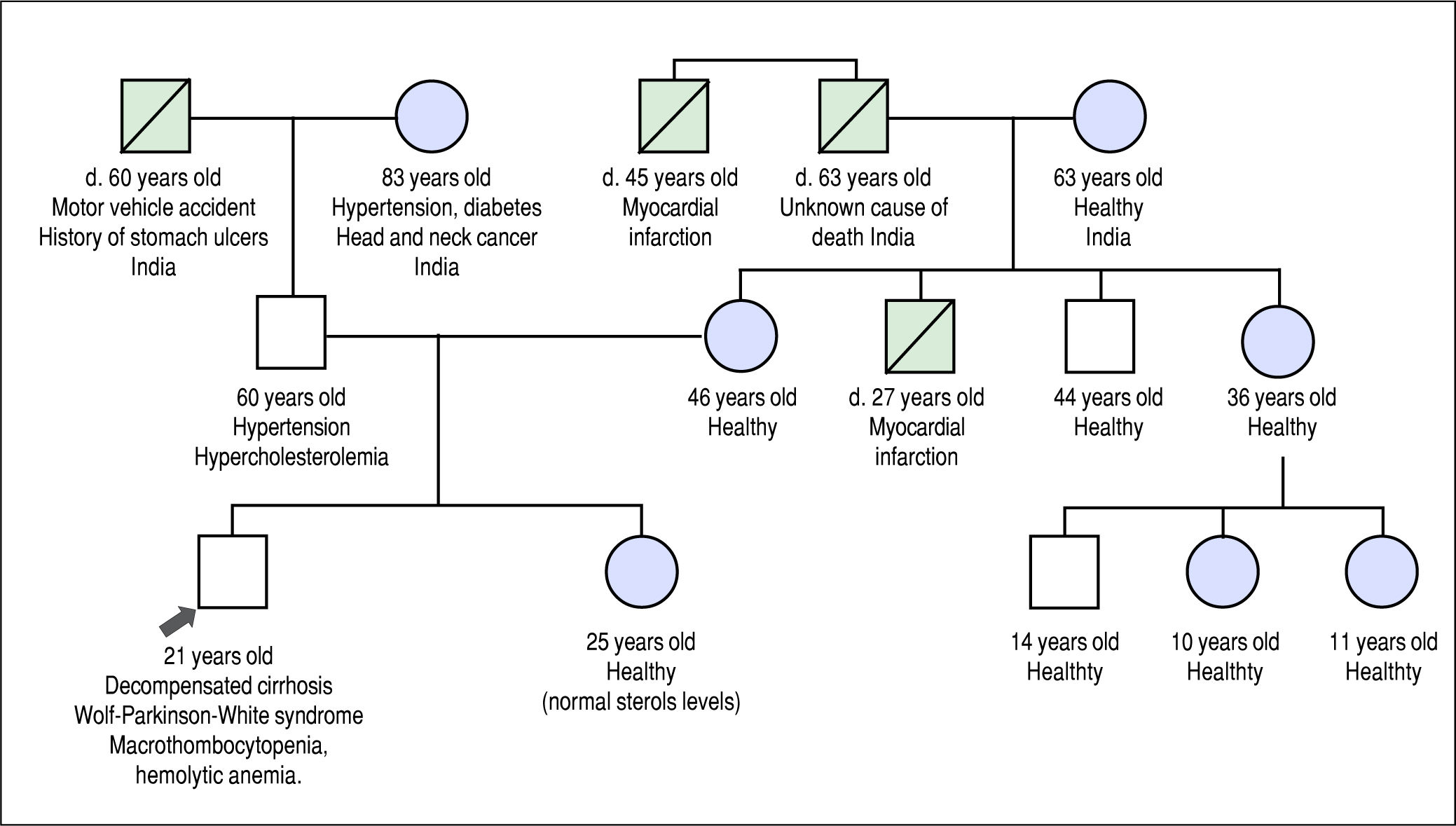
He was referred to our institution for a second opinion. His main symptoms were fatigue, unstable angina, and exer-tional dyspnea. Past history revealed that he experienced waxing and waning episodes of conjunctival icterus and change in urine color in childhood and adolescence. He was reportedly anemic during these episodes, but no further work-up was done. His daily dietary habits included consuming several portions of nuts, chocolate, fruits and vegetables per day, and he never consumed alcohol. His family history included premature death of a maternal uncle, thought to be related to a myocardial infarction at the age of 27 years, but was otherwise noncontributory (Figure 1).

Family history pedigree. Black arrow indicates the patient, age 21 years at diagnosis. Mother and father are apparently healthy. Maternal uncle died at 27 years of a presumed myocardial infarction. Maternal grand-uncle died of a presumed myocardial infarction at the age of 47 years.
Physical exam revealed a thin male in no distress. Scler-al icterus, diffuse jaundice, and several spider angiomata were noted. There was no jugular venous distention or gal-lop upon cardiac auscultation, and lungs were clear of crackles. Abdominal exam revealed large volume ascites. Musculoskeletal exam was notable for tendon xanthomas and bilateral Achilles tendon thickening. Neurologic exam demonstrated neither asterixis nor other focal deficit. Eye exam revealed no Kayser-Fleischer rings.
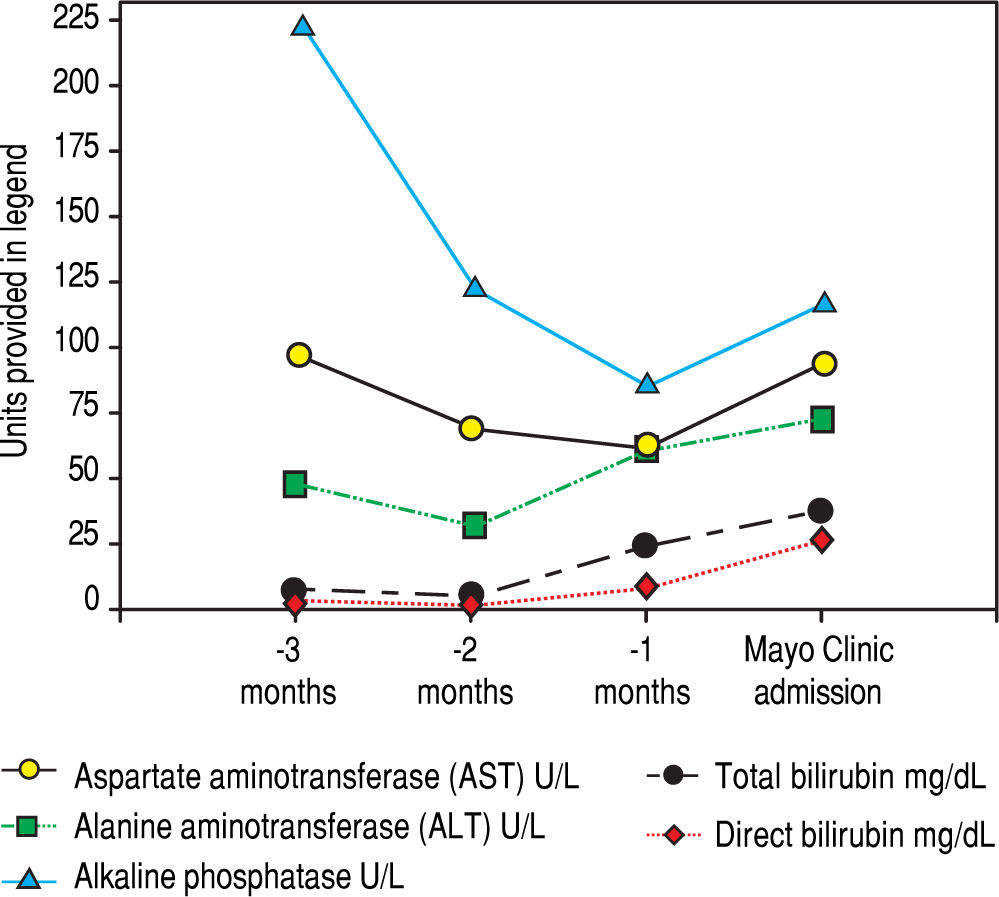
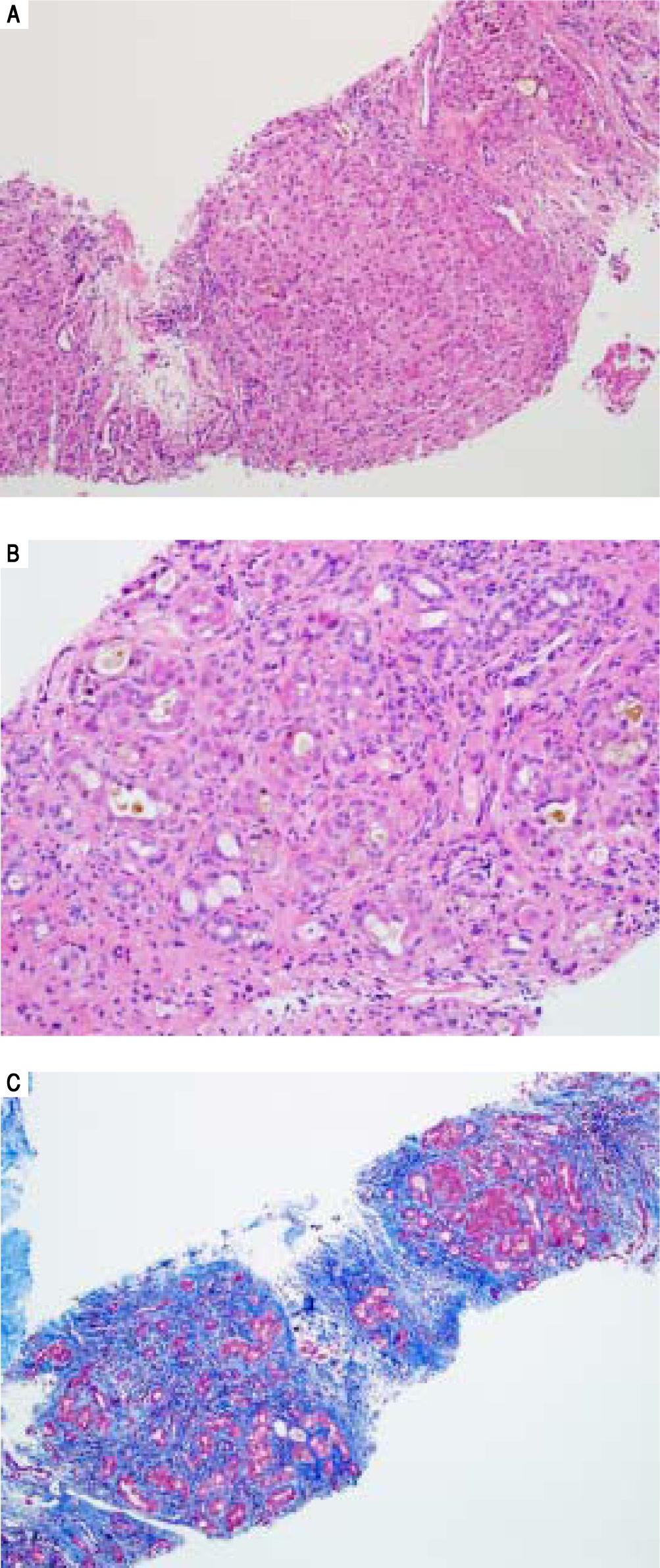
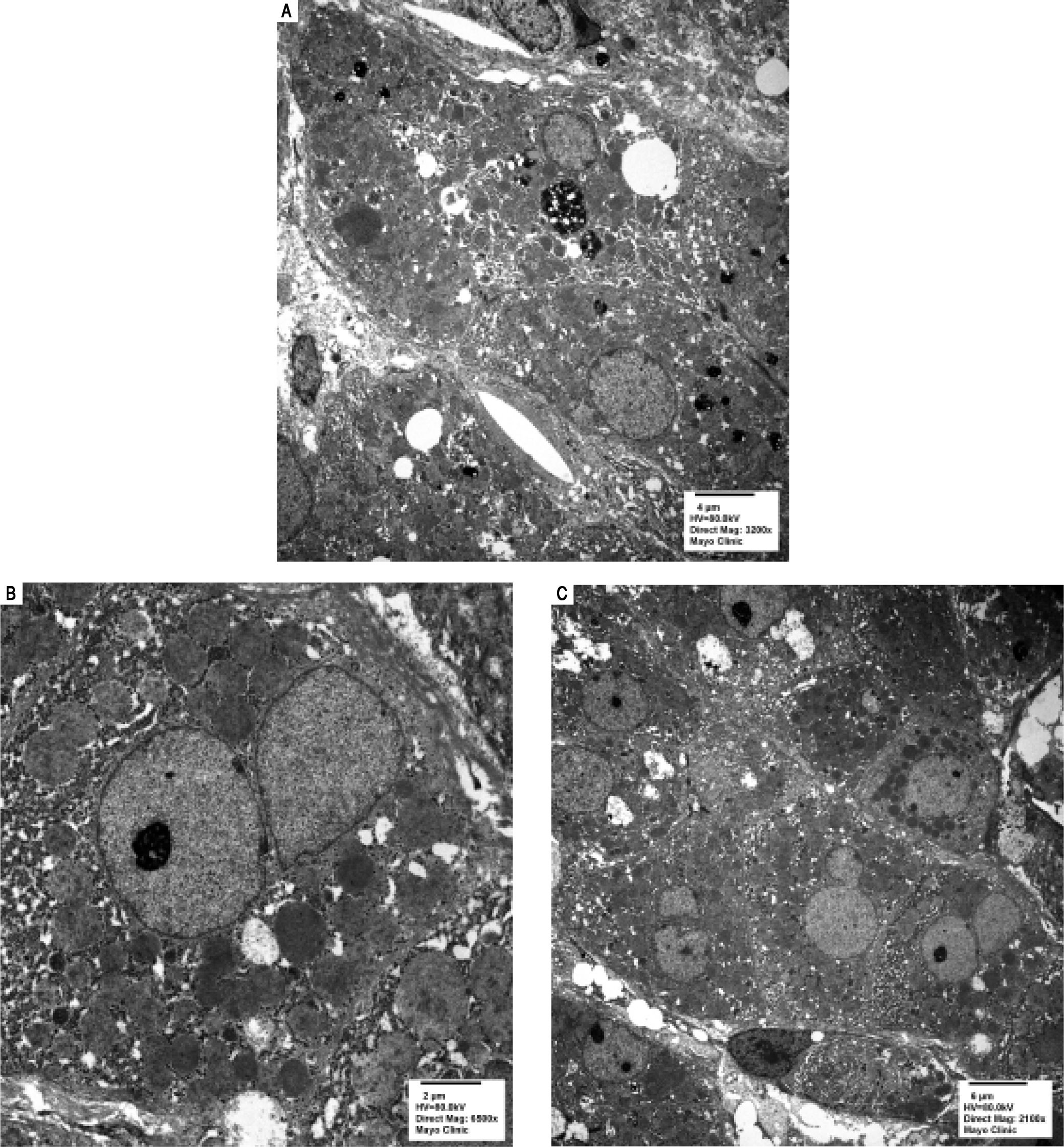
Serologic work-up ruled out chronic causes of liver disease, including negative hepatitis B and C serology, normal iron and copper studies, and negative auto-antibodies. On admission to our hospital, the patient's hemoglobin was 8.7 g/dL, with profound reticulocytosis (578 K/ mm3; 21% of red blood cells). Haptoglobin was undetect-able (although its interpretation was confounded by liver disease), and lactate dehydrogenase level was 540 U/L (normal < 222 U/L) without evidence of disseminated in-travascular coagulation on peripheral smear (only showed rare large platelets and thrombocytopenia). Polyspecific direct antiglobulin test and the Donath-Landsteiner test were negative, and flow cytometry ruled out paroxysmal nocturnal hemoglobinuria. Since the patient's blood pressure and serum electrolytes were normal, adrenal insufficiency was not evaluated. The patient's extensive laboratory work-up, as well as liver enzymes trend until admission to the Mayo Clinic, are noted in table 1 and figure 2. Histologic examination of a transjugular liver biopsy showed established cirrhosis (Figures 3A and 3C). The hepatic acinar parenchyma also displayed marked ca-nalicular and hepatocellular cholestasis (Figure 3B). There were occasional foci of hepatocellular parenchymal collapse. Electron microscopy confirmed the histologic finding of cholestasis and showed some increase both in the number and size of mitochondria (Figures 4A and 4B). Some increase in hepatic stellate cells was also identified (Figure 4C). No abnormal storage material was noted in hepatocytes or Kupffer cells.
Laboratory work-up on admission to Mayo Clinic.
| Hemoglobin 8.9 mg/dL. |
| MCV 106.1 fL. |
| Platelets 55 109/L. |
| Iron 121 mcg/dL. |
| Ferritin 232 mcg/L. |
| Iron saturation 64%. |
| Sodium 128 mmol/L. |
| Creatinine 0.6 mg/dL. |
| Thyroid stimulating hormone (TSH) 0.9 mIU/L. |
| Alkaline phosphatase 117 U/L. |
| AST 94 U/L, ALT 73 U/L. |
| Total bilirubin 38 mg/dL (direct bilirubin 26.4 mg/dL). |
| Gamma-glutamyl transferase 18 U/L. |
| INR 2.7 |
| MELD-Na Score= 34 points. |
| Urine excretion of Erythritol 54 mmol/mol Cr. |
| Urine excretion of Arabinitol 26 mmol/mol Cr. |
| Urine excretion of Ribitol 8 mmol/mol Cr. |
| Urine excretion of Sedoheptulose 10 mmol/mol Cr. |
| Homocysteine level 13 mcmol/L (normal). |
| Immunoglobulin G 3051 mg/dL.Anti-Smooth Muscle Antibody, Antimitochondrial Antibody, Liver/kidney microsomal antibodies, Antinuclear Antibodies, c-ANCA, p- ANCA, Antiphospholipids (IgM and IgG), Myeloperoxidase antibodies, proteinase (PR3) all negative. Alpha-1 antitrypsin level 121 mg/dL with MM phenotype. |
| Ceruloplasmin 19.2 mg/dL with normal 24 hour urine copper. |
| Evaluation for Cytomegalovirus (CMV), Epstein-Barr Virus (EBV), Human Immunodeficiency Virus (HIV), Herpes simplex viruses (type 1 and 2), Parvovirus, Hepatitis B virus (HBV), Hepatitis C virus (HCV), and Hepatitis A virus (HAV) was negative. Toxoplasma and syphilis evaluation were negative. |
| Acid-beta glucosidase > 38 nmol/mL/h. Sphingomyelinase 11.7 nmol/mL/h. Acid-alpha glucosidase 6.9 nmol/mL/h. Galactocerebrosidase 9.4 nmol/mL/h. Alpha galactosidase >20 nmol/mL/h. Alpha L-Iduronidase 5.9 nmol/mL/h. C20 Lysophosphatidylcholine 0.08 mcg/L. C22 lysophosphatidylcholine 0.07 mcg/L. C24 Lysophosphatidylcholine 0.06 mcg/L. C26 lysophosphatidylcholine 0.08 mcg/L. |
| Protein electrophoresis showed polyclonal hypergamma-blobulinemia: |
| Total protein 6.9 g/dL. Albumin 3.4 g/dL. Alpha-1 globulin 0.2 g/dL. Alpha-2 globulin 0.6 g/dL. Beta globulin 0.5 g/dL. Gamma globulin 2.3 g/dL. |
| Comprehensive urine toxicology screening was negative. |
| Bone marrow biopsy revealed normocellularity with erythroid hyperplasia, moderately-reduced granulopoie-sis, and normal megakaryopoiesis. There were no morphologic features of a myeloid neoplasm or involvement by a lymphoproliferative disorder. |

 stained with hematoxylin and eosin (H&E) staining showing cirrhosis. B. High-power (200x) stained with hematoxylin and eosin (H&E) demonstrating cholestasis. C. Low-power (100x) Masson")
: Cholestatic pigment and occasional lipid droplets. B. Magnification (6,500x): Increased and enlarged mitochondria. C. Magnification (2,100x): Prominent hepatic stellate cells.")
Additional laboratory investigations revealed a normal lipid pattern, with a total cholesterol level of 152 mg/dL.
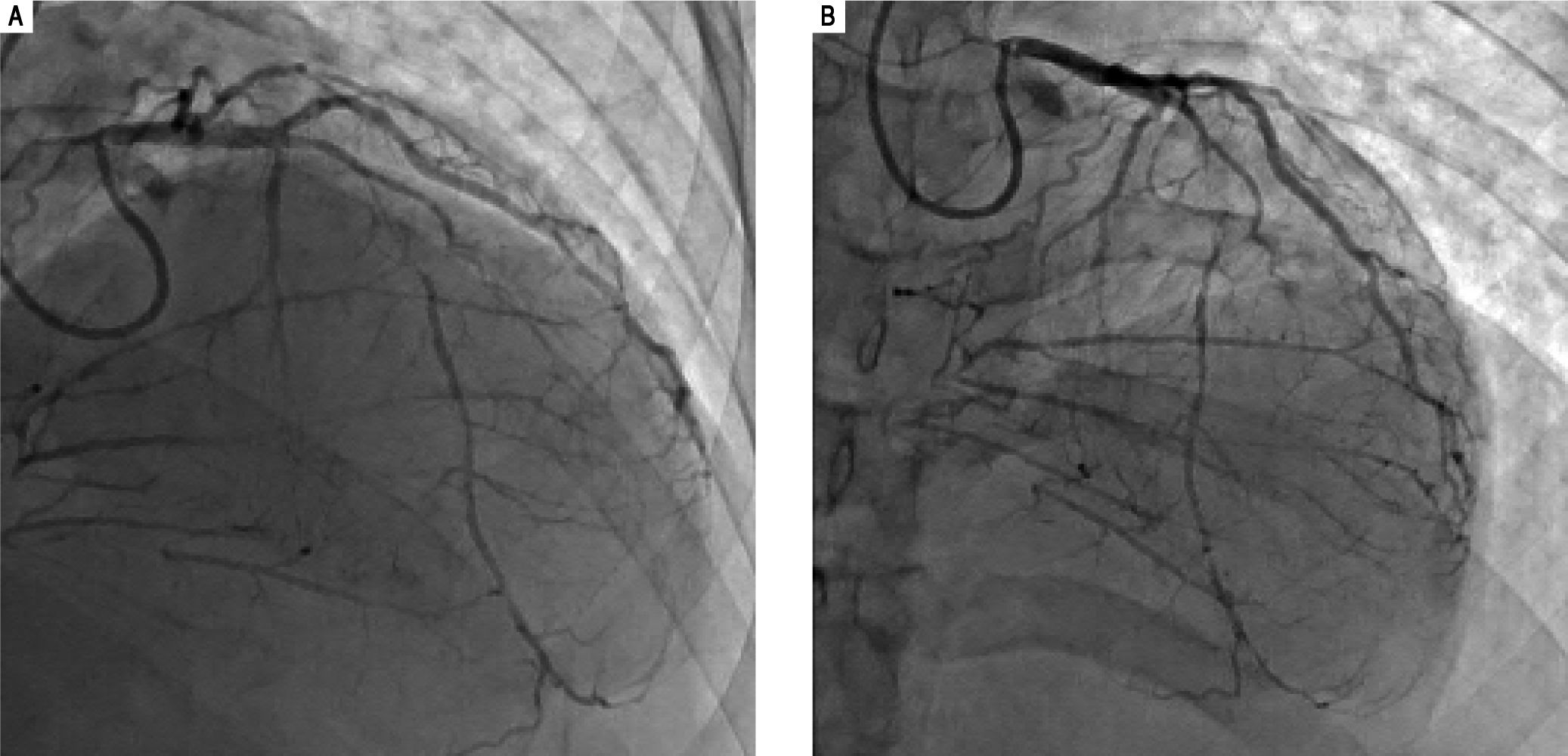
His outside coronary CT angiography showed complete occlusion of the proximal right coronary artery and segmental occlusions involving the medial segment of the left anterior descending artery. There were areas of collat-eralization, suggesting that his cardiac disease was chronic (Figure 5). Furthermore, atherosclerotic plaques, advanced for the patient's age, were noted in the abdominal and infra-renal aorta. We suspected that these lesions could represent accelerated atherosclerotic disease. Interestingly, no similar changes in the thoracic aorta were present, counter to what would be expected in a patient with extensive coronary atherosclerotic disease. Right heart catheterization revealed normal filling pressures, and slightly increased cardiac output.

Coronary angiogram images. A-B. Right and left anterior oblique cranial views, respectively, of the diffusely stenosed left anterior descending coronary artery, as compared with a relatively normal diagonal branch, and demonstrating significant collaterals that suggests an atretic right coronary artery.
In view of the tendon xanthomas, normal cholesterol levels, profound hemolytic anemia and diffuse coronary atherosclerosis, sitosterolemia was suspected. Plasma phy-tosterol levels were evaluated using gas chromatography-mass spectrometry (GC-MS)/GC-flame ionization detection (GC-FID). Sitosterol was elevated at 17.5 mg/ dL (normal < 0.5 mg/dL) as was campesterol at 7.9 mg/dL (normal <0.7 mg/dL). Although a biochemical elevation of phytosterols is sufficient to establish the diagnosis of si-tosterolemia, we opted to further confirm genetic mutations after a discussion with the patient and his family. This testing was performed on peripheral blood DNA at Fulgent Diagnostics Laboratory, and consisted of an interrogation of the ABCG5 and ABCG8 genes exons using next generation sequencing technology. The patient carried a homozygous pathogenic mutation in ABCG5 at c.1336C > T, which causes a premature stop codon at ami-no acid position 446, leading to a predicted truncated protein. No mutations in ABCG8 were present. Both parents, who did not undergo genetic testing, were first cousins and reportedly healthy.
Following this diagnosis, a low phytosterols diet and ezetimibe were initiated. He was approved for combined heart and liver transplant, given the ability to provide remission of sitosterolemia after transplant.8 Unfortunately, the patient passed away in the critical care unit due to a spontaneous intracranial bleed, while waiting for organ availability.
DiscussionOur patient was initially diagnosed with autoimmune hepatitis and cirrhosis with a loss of histological hallmarks of causative disease, similar to the case reported by Miet-tenen, et al.8 With a high level of clinical suspicion and appropriate laboratory investigations, sitosterolemia was diagnosed. Miettenen and colleagues were the first to report cirrhosis as the primary presenting feature in a sito-sterolemic patient, making our case the second in literature, and suggesting that this association is not coincidental. The known classic features of sitosterolemia are the presence of xanthomas despite normal cholesterol levels, macrothrombocytopenia, hemolytic anemia and premature atherosclerotic disease (Table 2). Prior to this report, end-stage liver disease has not been recognized as a cardinal feature of sitosterolemia, and we believe that our observation and review are pivotal in raising awareness to this lethal, and potentially reversible disease. The clinical presentations are variable and range from range from being asymptomatic to severe. Hemolysis is a frequent occurrence in affected individuals and is thought to be related to the effect of sitosterol on red cell membrane stability, and this feature may be episodic, interrupting periods of nor-mality.9 The presence of tendon xanthomas without elevated LDL-cholesterol should raise suspicion of sitosterolemia (grade III/B recommendation).10 Adrenal insufficiency,11 spinal cord compression12 and splenomeg-aly13,14 are also reported associations.
The range of clinical features currently seen in sitosterolemia compared to the clinical features of our patient.
| Clinical feature | Relative frequency in literature | Presence in our case |
|---|---|---|
| Arthralgia | ~30% | Yes |
| Tendon xanthomas | ~30% | Yes (bilateral thickening of Achilles tendons on ultrasound) |
| Elevated total cholesterol (mild) | ~30% | No |
| Anemia (mild) | ~30% | Yes (Severe) |
| Thrombocytopenia | ~30% | Yes |
| Elevated liver enzymes (<3 upper limit of normal) | ~30% | Yes |
| Thickened cardiac valves | < 10% | No |
| Carotid bruit | < 10% | No (No stenosis on carotid ultrasound) |
| Premature coronary disease | < 10% | Yes (Severe and diffuse) |
| Sudden premature death | < 10% | Yes (Due to spontaneous intracerebral bleed and multiorgan failure, not myocardial infarction) |
| Liver cirrhosis | < 1% | Yes (second known reported case) |
| Diarrhea (ulcerative colitis) | < 1% | Unknown but possible (patient hadan episode of bloody diarrhea priorto admission) |
| Vitamin D deficiency in childhood | Not reported | Yes |
| Wolff-Parkinson-White | Not reported | Yes |
| Nephrolithiasis | Not reported | Yes |
Biliary and enterocyte sterol handling is critical for the excretion of cholesterol, as well as non-cholesterols sterols, such as phytosterols.15 To date, the ABCG5/ABCG8 transporter apparatus is the only recognized system to participate in the process of phytosterol excretion. ABCG5 and ABCG8 form heterodimer transporters that are located in the apical cell membranes of the bile ducts and intestinal enterocytes. Sitosterolemia patients have biallelic pathogenic mutations in the genes, ABCG5 or ABCG8, causing loss of function of the ABCG5/ABCG8 transporters,5-7 which causes severe elevation in plasma phytosterols,
This accumulation of phytosterols could potentially lead to liver dysfunction phenotype of sitosterolemia. An association between sitosterol levels and cholestatic liver disease has been found in neonates who were administered sitosterol-containing lipid emulsions for TPN with increased incidence in a dose-dependent fashion. Kurvin-en, et al. concluded that elevated phytosterols, especially stigmasterol, may be an independent risk factor for liver disease in these patients.16 Similarly, in a more recent study by Hukkinen, et al., the authors found that among children with intestinal failure, parenteral plant sterols accumulate in the liver and relate to hepatic injury, portal inflammation, and liver fibrosis. The authors proposed a role for sterols in promoting liver damage.17
The underlying genetic alterations leading to sitostero-lemia are important to consider, as it delineates the fundamental underlying cause of the disease. Our patient had a homozygous variant in the ABCG5 gene (NM_022436.2) c.1336C > T. At the protein level, the nonsense variation p.Arg446* encodes for a premature stop codon at position 446, leading to protein truncation. In the other reported case of sitosterolemia-associated cirrhosis, the patient had a mutation in ABCG8, making this the first case of sitostero-lemia-associated cirrhosis in a patient with ABCG5 gene. To support the pathogenicity of this variation, there are two other patients with the same homozygous alteration diagnosed with sitosterolemia,18,19 although liver disease was not described. In the Exome Aggregation Consortium (ExAC) database, this variation is reported in 8 of 15,882 healthy individuals with an allele frequency of 0.0005%, further supporting evidence that this pathogenic mutation has a frequency in the general population consistent with a recessive condition (accessed on 10/6/2016).20
Sitosterolemia is thought to be a rare disease, however the true prevalence is unknown; the current estimation is 1 in 1,000,000.21 A Chinese group reported 13 patients from 8 separate families over a seven year period who presented with sitosterolemia to a single insti-tution.22 Therefore, it seems that sitosterolemia may not be as rare as originally thought, and we believe that screening is warranted in patients presenting with cryp-togenic cirrhosis with any cardinal manifestation, and may even be important before symptoms manifest altogether, considering the simple effective interventions (e.g. diet modifications, ezetimibe treatment) that may alter a sinister end-stage outcome. Although the relationship between sitosterolemia and liver disease has not been previously firmly established, this feature of clinical variability, termed variable expressivity, is common in many genetic disorders, including inborn errors of metabolism.23 In addition, this tenet of variable expressivity could account for the variability of clinical features in this disease. Our case suggests that the progressive liver dysfunction phenotype association is real and requires further study. A high index of suspicion is needed to make the diagnosis, as standard lipid testing does not identify this disorder, and phytosterols specific evaluation must be performed [Gas chromatography (GC) or high-performance liquid chromatography (HPLC)] separation of sterols. Alternatively, patients who are diagnosed with sitosterolemia should undergo a hepatology evaluation to determine the extent of liver involvement, and interval liver follow-up should be pursued. This is mainly directed at evaluating the presence of increased liver enzymes (alanine aminotrans-ferase, aspartate aminotransferase, alkaline phosphatase, and gamma glutamyl transferase), the integrity of the hepatic functions (e.g. prothrombin time, bilirubin, and albumin), as well as liver stiffness measurement as a surrogate for liver fibrosis (e.g. transient elastography). This will allow for the evaluation of the presence, development, and progression of hepatic affliction, which may modify the aggressiveness of sitosterolemia therapy. Furthermore, this will facilitate the recognition of end stage liver disease, and appropriate referral to a liver transplant center prior to decompensation, if needed.
This case presents novel and important observations regarding this possibly under-recognized disease; idio-pathic/crytogenic cirrhosis is not an uncommon diagnosis for some end-stage liver disease subjects. The association of cirrhosis and ABCG5 mutation has not been previously described. Miettenen, et al. hypothesized that a mutation in either ABCG5 or ABCG8 may cause misfolded proteins in the liver to accumulate and induce injury. However, their immunohistochemistry data did not support that hypothesis. The mutation detected in our patient, c.1336C > T is theorized to result in nonsense mediated RNA decay, meaning that the RNA would be targeted for degradation before translation occurs.24 As a result, we would hypothesize that staining for ABCG5 in liver tissue would be completely absent. Unfortunately, there was no tissue available to test for this possibility. In the only other reported case, liver transplantation resulted in significant amelioration in plant sterol values.8 Miettenen and colleagues hypothesized that transplantation of a nonsitosterolemic liver seems to prevent the accumulation of phytosterols, despite a persistent intestinal defect.8 The restoration of hepatobiliary excretion mechanisms remains a major pathway for sterol excretion and are sufficient to alleviate the phytosterolemia. In a mouse model of sitosterolemia, an intestinal-only restoration also seems to reduce the xenos-terol burden, suggesting any one pathway is sufficient to make an impact on the biochemistry.25
In summary, we report a novel case of sitosterolemia and cirrhosis, confirmed by biallelic mutations in ABCG5. We postulate that liver damage is likely secondary to the harmful effects of high plant sterols levels, with potential contribution from misfolded mutant protein accumulation in the hepatic cells. Increased awareness of the disorder is needed to identify new affected patients and establish more accurate frequency estimation of sitosterolemia. Consideration for phytosterol testing for patients presenting with cryptogenic cirrhosis, particularly if they display other cardinal features of sitosterolemia should take place. A thorough hepatology evaluation for those who are newly diagnosed with sito-sterolemia should also be performed, to monitor for the occurrence, as well as progression of liver disease. The magnitude of noble organ damage should guide the escalation of therapy, starting from dietary modification and ezetimibe use,2,26 to surgical ileal bypass, and perhaps liver transplantation, if ongoing hepatic damage is not subdued with noninvaive approaches.
AcknowledgementsWe are grateful to Dr. Lisa Schimmenti and Dr. Robert Steiner for their helpful and informative discussions regarding this case, to Dr. Ian Cheng-Yi Chang for reviewing our coronary angiography images, and to Dr. Samir Haffar for his review of the manuscript.
Financial Support and Conflict of Interest DisclosureNo financial support or conflict of interest for any of the authors is present.



