



The understanding of the mechanisms for the development of ascites has evolved over the years, involving the liver, peritoneum, heart, and kidneys as key responsible for its formation. In this article, we review the pathophysiology of ascites formation, introducing the role of the intestine as a major responsible for ascites production through “a game changer” case.
Ascites is a prominent clinical feature of cirrhosis. Although multifactorial, the main cause is altered portal hemodynamics. Portal pressure was first measured in 1896 [1], and the first human study took place in 1937 during a surgical procedure; since then, intra-splenic pressure measurement, umbilical vein catheterization, percutaneous trans-hepatic and hepatic vein wedge pressure have been utilized to document it [1,2]. In non-cirrhotic livers, there is a complex interplay between the portal vein and hepatic artery blood flow because of the hepatic arterial buffer response that occurs after a decrease in the portal flow [3]. Consensus has established that portal hypertension (PHT) is secondary to increased resistance to the blood flow in the hepatic veins, sinusoids, and/or the portal vein, as well as a subsequent development of increased arterial flow [1–9]. The liver has been considered the main source of ascites formation, and its development was read as the result of fluid leakage through the Glisson's capsule, mainly when right heart failure, Budd-Chiari syndrome, or cirrhosis, occur [7,8]. Diseases of the heart, peritoneum, and kidney can also contribute to, or be responsible for the development of ascites. Clinically, the priority in the management of PHT remains the avoidance and early treatment of potentially life-threatening complications such as upper gastrointestinal bleeding, encephalopathy, or spontaneous bacterial peritonitis. Ascites can be mostly managed pharmacologically, although in some cases, surgery remains a valuable alternative [1–11]. It has been noticed that patients suffering from cirrhosis and short bowel syndrome, whose liver disease is mainly related to chronic use of parenteral nutrition (PN) in the context of intestinal failure (IF), rarely produce ascites despite the presence of progressive PHT [8–13]. The comprehensive multidisciplinary team care of those patients triggered the need to better describe the role of the intestine and the intestinal circulation in the pathophysiology of ascites formation.
We herein present a concise review of different settings in which it can be highlighted the role of the intestine in the production of ascites, even in the absence of liver disease, and exemplifying the concept through a unique case of isolated superior mesenteric vein thrombosis.
2The clinical findings of liver disease as a consequence of using parenteral nutritionIn 1965, Lawson L. published a manuscript describing the use of PN in immediate post-surgical ICU care; he showed the effect of providing 2600 to 3000 Kcal/day, documenting for the first time the occurrence of jaundice and liver fibrosis in biopsies. In 1968, Dudrick SJ, Wilmore DW, Vars HM, Rhoads JE set the basis for using PN chronically in children and became one of the four major contributions to modern surgery after the use of penicillin, anesthesia, and antisepsis. The advances made by Scribner B. and Jeejeebhoy K. under the concept of “the artificial gut” project, allowed patients to go home on PN; this achievement was favored by the development of the tunneled central venous catheters by Broviack and Hickman.
The improvements and advances in the IF field increased patient survival and expanded the population of chronic home PN. This led to a greater number of IF-related complications, progressive liver disease is one of them. Intestinal failure-associated liver disease (IFALD) was, for years, the main indication for isolated or combined liver-intestinal transplantation. Patients with IFALD usually have hypersplenism and splenomegaly as the main manifestations of progressive liver fibrosis. Peristomal bleeding and coagulopathy are signs of liver failure in this patient population. Nevertheless, ascites is seldom observed in the setting of IFALD and short bowel; if ascites is present, there is longer residual intestinal length with the concomitant splanchnic circulation.
3The clinical manifestations of the porto-mesenteric vein thrombosisAscites is present in the setting of acute or chronic diffuse porto-mesenteric vein thrombosis, independently of the degree of liver disease. Diffuse thrombosis can occur as a consequence of cirrhosis of any etiology, as well as in patients with thrombotic disorders despite having a normal liver function. In the acute setting, the dominant life-threatening complication is variceal bleeding. In such cases, the intestinal length remains invariable, except in cases requiring partial or subtotal enterectomy.
When thrombosis occurs as part of a chronic and progressive disorder, ascites and progressive intestinal insufficiency/failure become the main clinical manifestations, leading in some cases, to intestinal failure.
When portomesenteric thrombosis occurs and is timely diagnosed, it mandates a shunt surgery to reduce the portal pressure; one of the most relevant contributions of the last years was the meso‑Rex bypass described by Superina R. et al. [14], which returns the splanchnic flow to the intrahepatic circulation. Bleeding and hypersplenism, followed by encephalopathy, are the principal symptoms described, attributing the development of ascites just to fluid resuscitation without considering any role to the intestines. In cases of advanced thrombosis (Grade IV), the only alternative is a multivisceral transplant, as it was shown by Vianna R. et al. in 2012. In this setting, 25 patients with porto-mesenteric vein thrombosis received 25 multivisceral transplants, obtaining at the 5-year patient and graft survival of 72%, reversing portal hypertension and addressing the primary disease in most cases [15].
4The clinical manifestations and possible therapies for isolated mesenteric venous thrombosisA case of isolated superior mesenteric vein (SMV) thrombosis, its clinical course, and the treatment implemented triggered our interest in writing this review and became the "proof of the concept" that the intestine plays a major role in the production of ascites
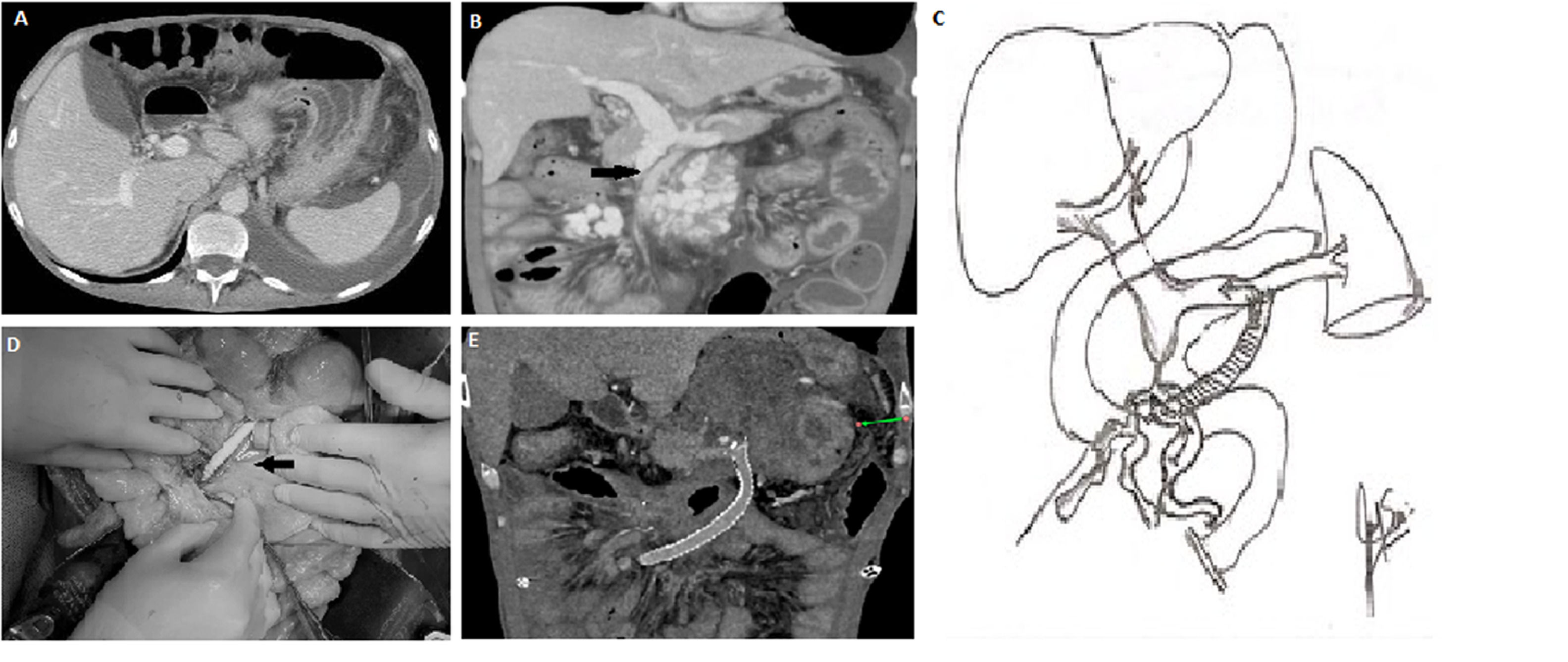
In 2018, a 39-year-old male with a past medical history of HIV developed ascites, diarrhea, and enlarged mesenteric lymph nodes seen on a contrast-enhanced abdominal CT scan. An exploratory laparotomy and lymph node biopsy confirmed the diagnosis of active tuberculosis. Treatment was initiated with isoniazid, ethambutol, and rifampicin for 12 months, in addition to the standard HIV therapy. Tuberculosis and HIV replication were controlled, achieving a negative viral load (<20 copies) and CD4 count of 353/mm3. During the hospitalization, he developed deep venous thrombosis of the left femoral, external, and common iliac veins; intravenous anticoagulation was initiated, followed by oral anticoagulation. In 2020, he started having chronic diarrhea and significant weight loss (13 kg over four months), necessitating PN as diarrhea persisted; an abdominal ultrasound, followed by a CT scan, confirmed the presence of a mesenteric vein thrombosis with the secondary development of significant collaterals in the mesenteric root. No splenomegaly was found, and splenic and portal vein flow through the liver was maintained (Fig. 1a). Over time, the diarrhea worsened. During the following 18 months and due to the use of a tunneled access for home PN, the patient developed right jugular and innominate venous truck thrombosis despite chronic anticoagulation. In June 2023, he was referred to our center for management. Physical examination was remarkable for thoracic and abdominal wall collaterals, as well as moderate ascites. His-weight was 47 kg, and his height was 172 cm (BMI: 15.9). His-blood chemistries were: Hct: 35.5%, Hb: 11.3 gr/dl., white blood cells cell count: 10.4 mm3, Platelet count: 420.000 mm3, Glucose: 88 mg/dl, Creatinine 0.71 mg/dl, sodium: 135, mEq/L, potassium: 3.3 mEq/L, aspartate aminotransferases: 17 UI/l, alanine aminotransferase: 19 UI/l, total bilirubin: 0.24 mg/dl, alkaline phosphatase: 127 U/l; total proteins were < 2.5 gr/dl. The analysis of the ascitic fluid was remarkable for white blood cell count <500 l, <50% neutrophils, LDH: 225 U/l, glucose: 70 mg/dl, total protein <1 g/dl, triglycerides: 418 mg/dl; the serum- ascites albumin gradient (SAAG) was 2.5. An abdominal CT scan confirmed (Fig. 1b) patent splenic and portal veins, absence of splenomegaly or hepatomegaly, chronic thrombosis of the SMV up to 3 mm below the splenic-portal confluence with large collateral circulation in the mesenteric root below the inferior edge of the pancreas. Moderate ascites and significant intestinal wall edema involving the small bowel and right colon were also noted. Thrombophilic and cardiovascular evaluation were normal.
 Preoperative CT scan: showing ascites, with normal size liver and spleen. B) Preoperative CT scan: showing SMV thromboses, SMV collaterals, intestinal wall edema and ascites, with preserved splenic vein and portal flow. C) Illustration of the SMV collateral to the splenic vein shunt. D) Surgical pictures of the shunt, showing in detail the SMV collateral anastomosis and the direction of the shunt towards the splenic vein anastomosis. E) Post-operative abdominal CT scan showing a patent shunt, absence of collaterals, intestinal wall edema and ascites. CT, computed tomography; SMV, superior mesenteric vein.")
A) Preoperative CT scan: showing ascites, with normal size liver and spleen.
B) Preoperative CT scan: showing SMV thromboses, SMV collaterals, intestinal wall edema and ascites, with preserved splenic vein and portal flow.
C) Illustration of the SMV collateral to the splenic vein shunt.
D) Surgical pictures of the shunt, showing in detail the SMV collateral anastomosis and the direction of the shunt towards the splenic vein anastomosis.
E) Post-operative abdominal CT scan showing a patent shunt, absence of collaterals, intestinal wall edema and ascites.
CT, computed tomography; SMV, superior mesenteric vein.
Isolated mesenteric vein thrombosis and regional venous hypertension causing ascites and progressive intestinal failure is a rare clinical picture. The normal liver function and vasculature and preserved splenic vein flow preclude the need for a multi-visceral transplant, but performing a total enterectomy followed by an isolated intestinal transplant might be considered if mesenteric thromboses expanded.
After a multidisciplinary team meeting, it was decided to perform a laparotomy to assess the pressure of the SMV territory and to perform a surgical shunt since TIPSS was not considered a possible option. At laparotomy, the liver and spleen had a normal gross appearance; a liver biopsy showed mild steatosis; 1.5 liters of chylous ascites were evacuated from the abdominal cavity. The visceral peritoneum was edematous, with an intestine grossly boggy with reduced motility.
The pressure of the SMV collaterals was found to be 30 mmHg. A prominent venous dilatation was identified at the root of the mesentery, close to the thrombosis at the infra-pancreatic SMV, and the infra-renal IVC could be approached just above the iliac veins. Then, a 6 mm ringed Gore-vascular graft was sutured to the SMV collateral vein and the distal end to the lower IVC, and flow was established. The new measurement of the SMV system was 5 mmHg. Before closing, the small bowel edema started to improve, and the visceral motility improved. A heparin drip was initiated in the OR. On post-operative day 1, abdominal drains collected 650 mL of serous fluid. Abdominal ultrasonography showed turbulent flow in the IVC, which was an indirect sign of shunt patency. Similar findings were observed on post-op days 2 and 3, but on day four, an increase in the amount of abdominal fluid drainage was observed (up to 1500 mL). A contrast abdominal CT scan showed thrombosis of the shunt and a significant reduction in the lumen of the IVC at the level of the left renal vein, which was not identified before. The patient returned to the operating room for a second intervention. SMV pressure was reassessed as 35 mmHg. The previous shunt was removed. The retro-pancreatic splenic vein was identified, and a new 8 mm Gore-ex vascular-ringed graft was anastomosed to the same SMV collateral vein and the splenic vein. Once the flow was re-established, the SMV pressure decreased to 6 mmHg (Fig. 1c and d). A new contrast CT scan performed on post-op day 2 showed patency of the shunt was reassessed, and a follow-up CT scan was done a month later (Fig. 1e). The post-surgical serum-ascites albumin gradient (SAAG) was 1.1. No diuretics were used, and anti-coagulant therapy was continued. The patient was discharged without PN 21 days after the first procedure, with one abdominal drainage that was removed after one month. At present, four months after the surgery, the patient's weight increased to 58 kg, and all chest and abdominal wall collaterals disappeared. No ascites was found in a follow-up ultrasound, and the diarrhea was resolved.
To the best of our knowledge, neither a single case nor a small series of similar cases have been reported.
5The role of the intestine in the production of ascitesMesenteric thrombosis and splanchnic hypertension, strictly limited to the intestine, underline their central role in the pathogenesis of ascites. For many years, the intestine was a forgotten organ of the abdomen. Nevertheless, it plays a central physiologic role in the abdomen; it contains as many neurons as the spinal cord, it holds 80% of the total body lymphocytes, it produces several entero-hormones, and it participates not only in the digestion and absorption of food but also in many metabolic and hormonal pathways [16,17].
The possibility of having selective thrombosis of the mesenteric vein challenges the dogma that the liver, the heart, the kidney, or the peritoneum are the main responsible for the development of ascites [1–11,14,18,19].
The concept of the liver-intestinal axis requires to be broadened to better understand the pathophysiology of portal hypertension and ascites [12,13,16].
Portal hypertension is associated with increased resistance to portal blood flow; therefore, the increased pressure is conveyed directly not only to the spleen but also to the intestine, leading to portal hypertensive enteropathy. The chronically increased portal/splanchnic pressure and venous stasis affect the intestine, causing edema of the whole intestinal wall, featuring thick-walled dilated vessels along with edema of the lamina propria, fibromuscular proliferation, decreased villous/crypt ratio, and thickened muscularis mucosae, leading to compromised absorption and motility [8].
The increased intramural pressure also compromises the centripetal lymph flow, overloading the lymphatic fluid production; this would ultimately reverse the lymph flow towards the serosa, causing capillary and peritoneal leakage. The final expression of this impairment is obvious in this case, where the disruption of the lymphatic pathway produces chylous ascites [7,8,19–21].
This case allows broader considerations. First, it may well substantiate the origin of malnutrition in cirrhotic patients. Its prevalence has been reported to be highly variable (5–92%), and its cause is read as multifactorial, including impaired dietary intake, altered micro and macronutrient metabolism, energy metabolism disturbances, increased energy expenditure, impaired bile acid metabolism, gut microbiome dysregulation, and small intestinal bacterial overgrowth [20–22]. Nevertheless, intestinal wall edema and lymphatic leak might have a major responsibility in malnutrition by generating malabsorption. What is more, malnutrition resolves in cirrhotic patients when TIPS are placed [22,23] and following liver transplantation [24].
Secondly, the case suggests that pathophysiological mechanisms for spontaneous bacterial peritonitis could be the result of combining an increased mucosal permeability due to intestinal stasis with the reversed lymph flow toward the peritoneal cavity. Half of the lymph in the body originates in the intestine, and its leakage to the peritoneum favors bacterial translocation, leading to the development of spontaneous bacterial peritonitis [2,3,7]. In the setting of mesenteric fibrosis, in carcinoid tumors, for example, similar portal hypertension has been known to occur, supporting this possibility [25,26].
In addition, multiple reports proved the association between short bowel syndrome (mainly in ultrashort -<50 cm- intestine) and PN, in the development of progressive liver disease. However, patients with IFALD and cirrhosis do not develop ascites, albeit the most common manifestations of advanced liver disease in that setting are hypersplenism, progressive cholestasis, and peri‑stomal bleeding in the presence of any type of ostomy. This leads to a late diagnosis of IFALD, missing the earliest stages [27,28]. Autologous gastrointestinal reconstruction or isolated intestinal transplantation proved the reversibility of liver fibrosis once intestinal length is recovered and PN is discontinued [29], again demonstrating the close functional link between the two organs [27–32].
Finally, this case exemplifies the development of ascites in the absence of liver, peritoneal, renal, or cardiac disorders and only as a consequence of isolated SMV hypertension. The proportion of ascites caused or produced by the intestine, in the setting of the other mentioned disorders, requires being determined.
The treatment instituted in this case was based on the existence of what could be named: "pre-portal venous hypertension," following the classical classification of portal hypertension, and based on the location of the thromboses at the SMV outflow just before the junction with the splenic vein [14]. The reduction of the increased resistance to the hydrodynamic flow and pressure to the SMV system by shunting it into the splenic vein reverses the clinical picture. That change in the pressure of the splanchnic venous circulation will lead to the final resolution of the intestinal wall edema, chylous ascites, diarrhea, and finally, intestinal failure. In our case, three weeks after the shunt surgery were necessary to resolve all his years of suffering.
This novel shunt, SMV collateral to splenic vein, allowed the flow to be diverted into a lower-pressure venous system, requiring the use of a short vascular graft. In addition, the indirect aspirate effect produced by the diaphragmatic movements during the respiratory cycle benefits the blood flow into the portal system [33,34] Despite its ascendant and anterior-to-posterior placement.
6ConclusionsThis review aims to finally underline the value and brings a clearer understanding of the whole liver-intestine unit, pondering the role of the intestine in the overall pathophysiology of the gastrointestinal tract and the production of ascites.
Author contributionsGabriel Gondolesi: Conceptualization, article design, drafting, and final approval. Carolina Rumbo: Article drafting and critical review. Leonardo Montes: Data acquisition. Lucia Novellis: Data acquisition and manuscript drafting. Diego Ramisch: Data analysis and interpretation, article revision. Ariel Riquelme Henriquez: Data acquisition. Mariana Ortega: Data acquisition. Federico Viano: Data acquisition. Thomas Schiano: Conceptualization, data analysis, and critical review. Valeria Descalzi: Manuscript drafting. Claudio Tiribelli: Conceptualization, data analysis, critical review, and final approval. Mihai Oltean: Conceptualization, data analysis, and critical review. Pablo Barros Schelotto: Manuscript revision and critical review. Hector Solar: Manuscript drafting.



