






The liver is the largest parenchymatous organ of the body and central for intermediate metabolism. Its close contact to the portal blood streaming back from the gut and the systemic circulation is a prerequisite for the various processes ongoing on cellular level. The spatial relationship of the hepatocytes with the different cellular structures of the liver, such as fenestrated sinusoidal endothelial cells, Kupffer cells, and bile canaliculi is important for the organization and performance of the intermediate metabolism of nutrients (proteins, glucose and fatty acids), for the clearance of toxic or infectious agents, for metabolic detoxification and for the excretion of waste products.
The administration of chemotherapy is a challenge for the tight regulation and balance of these processes. As most drugs tend to be lipophilic, they are readily taken up by the liver. Under chemotherapy, up to 85% of patients develop liver steatosis. Steatohepatitis is the more serious event, especially if accompanied by an increase in bilirubin levels.
Modern understanding of the efficacy, safety and tolerability of combination chemotherapy has to increasingly include the individual context of a patient, such as age, gender, nutritional status, underlying diseases, genetic predisposition, as well as the cross-reactivity of the different drugs.
This review tries to capture the various effects of chemotherapy on the liver, with a focus on chemothe-rapeutical compounds used for the treatment of gastrointestinal cancers.
The liver is the central organ of the intermediate metabolism and is the main source of plasma proteins (e. g. albumin). The parenchymal cells, the hepatocytes, comprise about 80% of the liver mass and about 60% of the total number of the adult liver cell population.1,2 They are responsible for the organization and for the performance of intermediate metabolism, for the metabolic detoxification and for the excretion of waste products. Whilst for most drugs, hepatocyte function is drug-degradation, for some it is the activation of prodrugs. Therefore, a normal functioning hepatocyte is a prerequisite for the administration of chemotherapy. In case of abnormalities due to hepatocellular damage on the one hand, or to a high demand for hepatocellular biosynthesis on the other hand, this high-powered engine may get into trouble.
For the correct execution and tight regulation of these processes, the spatial relationship of the hepa-tocytes with the different cellular structures of the liver, such as fenestrated sinusoidal endothelial cells, Kupffer cells, and bile canaliculi is important.
Beneath the well known functions of the liver regarding intermediate metabolism and detoxification, there is another equally relevant function, the clea-rance function.3 About 80% of the body’s fixed tissue macrophages (von Kupffer cells) reside within the liver. The von Kupffer cells are in direct contact with the portal blood, streaming back from the gut or from the systemic circulation. This blood may be heavily loaded with toxic (lipopolysaccharide) or infectious agents (bacteria, viruses), cellular debris (e. g. from erythrocytes), undigested biomaterial or any other compounds that are not supposed to enter the systemic blood circulation. Scavenging of all this material is achieved in the liver by von Kupffer cell-and sinusoidal endothelial cell-endocytosis.4 Moreover, humoral factors that are part of the innate immune system may ease hepatic clearance and maintain host-defense mechanisms. Changes in the function of the liver immune cells have been reported after cytotoxic chemotherapy.
Alongside functional issues, obstruction of the bile duct system prior to chemotherapy presents a problem in cancer patients which has to be solved before chemotherapy is started (Figure I). Disturbance of the bile flow leads in fact to reduction of the hepatocellular function and to reduced protein synthesis. The gastroenterologist is therefore the specialist again, who should be contacted in such cases.
 showed a strong intrahepatic cholestasis with high bilirubin levels (E). Therefore a PTC was performed. The PTC (B) showed a dilation of the intrahepatic bile ducts and a subtotal stenosis of the DHC. In a “Rendezvous-technique” (PTC together with ERCP; C) it was possible to pass the DHC stenosis and place one stent in the right and one stent in the left main bile duct (D). The bilirubin levels decreased and a chemotherapy with gemcitabine was started. Actually the patient has further on normal bilirubin levels (E).")
A 62 years old patient with the first diagnosis of a cholangiocarcinoma in June 2006. The MRCP (A) showed a strong intrahepatic cholestasis with high bilirubin levels (E). Therefore a PTC was performed. The PTC (B) showed a dilation of the intrahepatic bile ducts and a subtotal stenosis of the DHC. In a “Rendezvous-technique” (PTC together with ERCP; C) it was possible to pass the DHC stenosis and place one stent in the right and one stent in the left main bile duct (D). The bilirubin levels decreased and a chemotherapy with gemcitabine was started. Actually the patient has further on normal bilirubin levels (E).
Chemotherapy as the main nonsurgical remedy of cancer treatment is generally based on the cytotoxic effect of natural or synthetic agents taking advantage of a higher vulnerability of cancer cells as compared to normal cells. However, normal cells may be affected by cytotoxic chemotherapy as well.5 Suscep-tibility to chemotherapy is linked to the proliferative rate of a cell population as seen in bone marrow cells or mucosal cells with high replication rates. Inhibition of DNA replication and transcription via interference of cancer drugs either with the DNA strand, with enzymes involved in DNA metabolism or DNA substrates may rank as the most important principle of cancer treatment directly inhibiting cell proliferation.
The Liver May Become A “Target” of and May Suffer From Cytotoxic ChemotherapyReplication of hepatocytes is low in normal liver, but may reach a high level during liver regeneration after massive hepatocellular death or partial hepa-tectomy.
In the healthy liver, with its low replication rate, inhibition of hepatocellular replication during chemotherapy thus is not of primary matter. Nevertheless, systemic application of chemotherapeutics affecting DNA, RNA or protein synthesis, may affect hepatocellular function in several ways:
Nausea and vomiting cause a reduction of nutrition; reduced uptake of nutrients has been shown to induce a shrinkage of the liver that may reach a reduction in liver cell mass of almost 50% under total starving. Normal food uptake after fasting has been shown to induce DNA synthesis in hepatocytes with cellular replication (DNA endoreplication) resulting in polyploidy and presence of several nuclei in the hepatocytes.6 However, even without a period of fas-ting, highly metabolically active hepatocytes need normal DNA and RNA synthesis for normal protein synthesis.7
Impact of Cytotoxic Chemotherapy on Liver Function: Hepatocellular Dysfunction and Clinical ImplicationsMost drugs tend to be lipophilic compounds that are taken up readily by the liver but that cannot be excreted easily unchanged in bile or urine. Metabolic pathways are inducible and include a series of steps that alter the parent molecule, synthesize conjugates of the drug or its metabolite with a more water-soluble moiety and comprise energy-dependent pathways to excrete either the parent molecule or its conjugates into the bile.8
Under chemotherapy, up to 85% of patients develop liver steatosis indicating disturbed lipid metabolism via altered lipoprotein synthesis in the hepato-cytes. An increase of the hepatocellular lipid content (Figure 2) is responsible for higher vulnerability. The higher vulnerability may, in particular during repeated chemotherapy, induce irreversible hepato-cellular damage through recruitment of inflammatory cells.

Elevation of serum aminotransferases represents a frequent event during or after cytotoxic chemotherapy. The hepatocellular sensibility to cytotoxic chemo-therapy depends on the particular chemotherapeutic agent, giving rise to a classification of agents with high or low potential hepatotoxicity (Table I).
Hepatotoxicity of cytotoxic chemotherapeutics.
| Hepatotoxicity | |
|---|---|
| Methotrexate | +++ |
| Asparaginase | ++ |
| Carmustine | ++ |
| Mercatopurin | ++ |
| Capecitabine | + |
| Chlorambucil | + |
| Cyclophosphamide | + |
| Cytarabine | + |
| Dacarbazine | + |
| Doxorubicin | + |
| Etoposide | + |
| Gemcitabine | + |
| Mitomycin C | + |
| Streptozocin | + |
| Busulfan | (+) |
| Cisplatin | (+) |
| 5-FU | (+) |
| Irinotecan | (+) |
| Imatinib | (+) |
| Oxaliplatin | (+) |
| Vincristine | (+) |
| Bevacizumab | 0 |
| Cetuximab | 0 |
| Epirubicin | 0 |
| Hydroxyurea | 0 |
| Rituximab | 0 |
+++ very often. ++ often. + rare. (+) very rare. 0 no hepato-toxicity
As described above, the liver is home of 80% of all fixed tissue macrophages of the body. It has to be assumed that this accumulation of mononuclear phagocytes in the liver reflects a high demand for clearance in this particular organ primarily conducted by Kupffer cells. The turnover of Kupffer cells has been debated for decades, and there is no final argument to what extent Kupffer cells are self-repli-cative or replaced by immigrating blood monocytes. The mitotic index of Kupffer cells is quite low, and increasing evidence has accumulated that Kupffer cells primarily derive from bone marrow cells.9 Accordingly, a numerous population of transient mo-nocytes has been reported in normal liver.10 On the other hand, a large influx of blood monocytes is observed in the diseased liver.11 After chemotherapy with suppression of bone marrow cell replication, the supply of Kupffer cell precursor cells is disturbed. At the same time, epithelial cell replication in the gut is also suppressed. The increased permeability of the gut gives rise to a highly increased hepatic onflow of toxic and infectious agents demanding a faster turnover of hepatic macrophages. This divergency may result in an increased risk for infections. It has further been shown that cytotoxic chemotherapy impairs the phagocytotic activity of Kupffer ce-lls.12 Reduced synthesis of humoral factors for host defense may also be critical in development of infections after cytotoxic chemotherapy.
Drug-Induced Liver DiseaseDrug-induced liver disease can mimic all forms of acute and chronic hepatobiliary diseases. Liver injury is designated hepatocellular when the alanine transaminase (ALT) level is greater than 2 times the upper limit of normal (ULN) or the ALT/alkaline phosphatase (AP) ratio is ≥ 5; cholestatic when the AP is greater than 2 times ULN or the ALT/AP ratio is ≤ 2; and mixed when the ALT/AP ratio is 2 to 5 and the individual values are greater than 2 times ULN.13,14
Hepatic dysfunction under chemotherapy mainly consists of abnormal biologic liver tests indicating chronic cholestasis with elevation in the levels of bilirubin, alkaline phosphatase (AP), and gamma glutamyl transferase (yGT) with or without abnormal levels of aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT). The predominant clinical presentations resemble acute icteric hepatitis or cholestatic disease.15
The development of hepatotoxicity is determined by the interplay of the toxic potential of the drug or its metabolites and the susceptibility of the patient as determined by genetic (i.e. extensive or poor me-tabolizer) and environmental factors.
Risk factors for drug-induced liver disease include: age, gender, genetic or familial predisposition, drug interactions (concomitant drugs), cross-reactivity of different drugs which have not been tolerated in the past, alcohol (abuse), nutritional status and underlying liver or other diseases (i.e. viral hepatitis).15 In cancer patients, causes of abnormal liver tests are further toxicity of anticancer drugs, toxicity of supportive medications, radiation-induced liver disease, tumor infiltration of the liver, Budd-Chiari syndrome, paraneoplatic syndrome, graft-versus-host disease (after stem cell transplantation), fungal liver disease, sepsis, and hemolysis.16
Veno-Occlusive Disease of the LiverHigh-dose cytoreductive chemotherapy (as given before bone-marrow transplantation) which might be combined with radiotherapy, carries the serious risk for the development of veno-occlusive disease (VOD) of the liver.17 The clinical hallmarks for the on-setting of this disease are jaundice, painful hepatomegaly and fluid retention, accompanied by high elevation of aminotransferases and later ascites and hepatic encephalopathy. The basic pathophysiological mechanism of induction of VOD of the liver is supposed to be an injury of the endothelial cells of central veins and sublobular liver veins.18 A subsequent clotting activation results in occlusion of central veins, sublobular veins and, in the worst case, of liver veins. Fibri-nolysis may have a positive effect on the outcome of this disease, and single cases of an emergency implantation of a transjugular portosystemic shunt (TIPS) have been reported.19 A final option for preventing the fatal outcome would be liver transplantation.20
Differential Diagnosis of Elevated Liver EnzymesDifferential diagnosis of elevated liver enzymes under chemotherapy has to include toxic effects from the tumor itself, resulting i.e. from hepatic me-tastases, tumor-induced portal vein thrombosis, pa-raneoplastic syndromes, infiltration with amyloid or light chain deposits or pre-existing liver disease, and furthermore effects from co-medication and infectio-ns.21
Causality assessment is necessary to determine the probability that liver injury is drug-induced. It includes the following steps:13,14
- •
Latency of liver-enzyme elevation following the administration of the drug.
- •
Rate of resolution after dechallenge.
- •
Risk factors for the development of liver disease (age, alcohol, pregnancy).
- •
Exclusion of other causes (viral hepatitis, ischemia, biliary tract disease, alcohol).
- •
Concomitant drugs.
- •
Track record of the patient’s charts/of similar cases.
- •
Rechallenge with the drug (in certain cases).
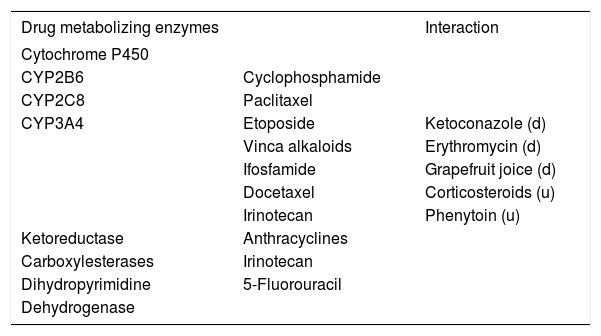
The degradation and detoxification of many che-motherapeutics in the liver depends on the activity of a particular enzyme pathway that may be altered by hepatocellular dysfunction induced by the drug itself.22 Moreover, genetic variations or inhibition of degrading enzymes by non-cytotoxic drugs may delay hepatic metabolization of the cytotoxic agent, thus increasing its toxicity.23 A contrary effect may be observed if the cytotoxic agent is in need of activation by a hepatic enzyme (pro-drugs), or if a drug-interaction increases the expression of a hepatic enzyme (Table 2).24 Impaired detoxification may be accompanied by disturbed biliary excretion of hepa-tocytes due to a chemotherapy-included slow-down of a the hepatocellular metabolism-inducing ele-vation of serum parameters of cholestasis.
Liver chemotherapeutics-metabolizing enzymes and interaction with drugs.
| Drug metabolizing enzymes | Interaction | |
|---|---|---|
| Cytochrome P450 | ||
| CYP2B6 | Cyclophosphamide | |
| CYP2C8 | Paclitaxel | |
| CYP3A4 | Etoposide | Ketoconazole (d) |
| Vinca alkaloids | Erythromycin (d) | |
| Ifosfamide | Grapefruit joice (d) | |
| Docetaxel | Corticosteroids (u) | |
| Irinotecan | Phenytoin (u) | |
| Ketoreductase | Anthracyclines | |
| Carboxylesterases | Irinotecan | |
| Dihydropyrimidine | 5-Fluorouracil | |
| Dehydrogenase |
d: downregulation. u: upregulation of the enzyme by the agent
Furthermore, enzyme polymorphisms of drug-metabolizing enzymes have to be taken into account, such as thiopurine S-methyltransferases (TMPT), di-hydropyrimidine dehydrogenase (DPD), aldehyde de-hydrogenases (ALDH), glutathione S-transferases (GST), uridine diphosphate glucuronosyltransferases (UGTs) and cytochrome P450 (CYP 450) enzymes.25
It has been shown, that genetic variants in the UDP-glucuronosyltransferase 1A1 predict the severe risk of neutropenia with irinotecan treatment.26–28 Lack of DPD results in severe 5-fluorouracil induced toxicity.29
For completion, some tumors are able to overex-press multidrug-resistance genes, resulting in poor responses of the tumor to chemotherapy.30
As a result evaluation of enzyme-polymorphisms as well as of drug-plasma levels is currently gaining increased importance for the improvement of indivi-dualized therapy.31,32
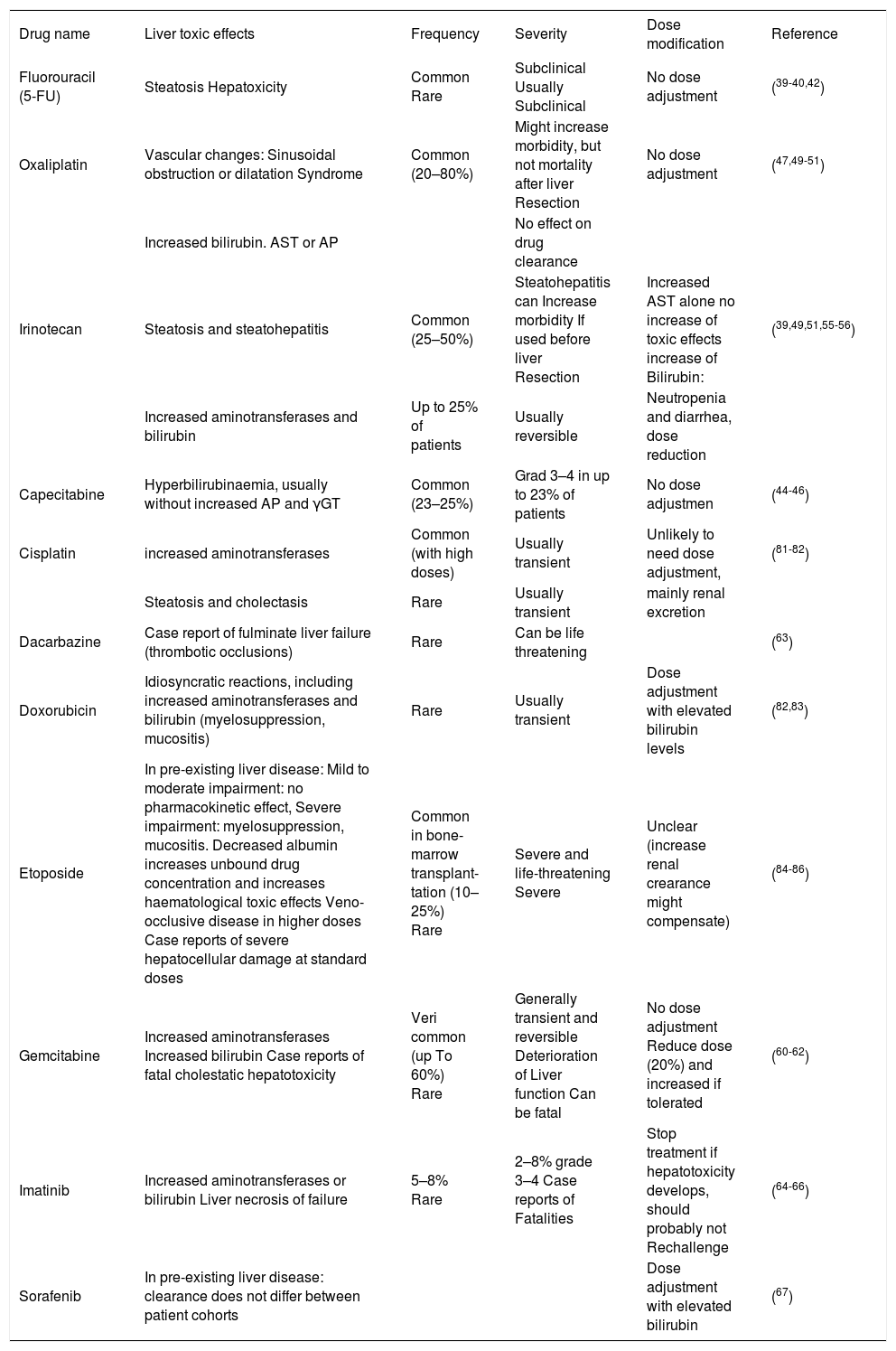
Liver Disease With Therapy Regimen Used for Gastrointestinal TumorsIn case of chemotherapy-induced hepatotoxicity, dose adjustment or change of the drug-combination may become necessary (Table 3).33–35
| Drug name | Liver toxic effects | Frequency | Severity | Dose modification | Reference |
|---|---|---|---|---|---|
| Fluorouracil (5-FU) | Steatosis Hepatoxicity | Common Rare | Subclinical Usually Subclinical | No dose adjustment | (39-40,42) |
| Oxaliplatin | Vascular changes: Sinusoidal obstruction or dilatation Syndrome | Common (20–80%) | Might increase morbidity, but not mortality after liver Resection | No dose adjustment | (47,49-51) |
| Increased bilirubin. AST or AP | No effect on drug clearance | ||||
| Irinotecan | Steatosis and steatohepatitis | Common (25–50%) | Steatohepatitis can Increase morbidity If used before liver Resection | Increased AST alone no increase of toxic effects increase of Bilirubin: | (39,49,51,55-56) |
| Increased aminotransferases and bilirubin | Up to 25% of patients | Usually reversible | Neutropenia and diarrhea, dose reduction | ||
| Capecitabine | Hyperbilirubinaemia, usually without increased AP and γGT | Common (23–25%) | Grad 3–4 in up to 23% of patients | No dose adjustmen | (44-46) |
| Cisplatin | increased aminotransferases | Common (with high doses) | Usually transient | Unlikely to need dose adjustment, | (81-82) |
| Steatosis and cholectasis | Rare | Usually transient | mainly renal excretion | ||
| Dacarbazine | Case report of fulminate liver failure (thrombotic occlusions) | Rare | Can be life threatening | (63) | |
| Doxorubicin | Idiosyncratic reactions, including increased aminotransferases and bilirubin (myelosuppression, mucositis) | Rare | Usually transient | Dose adjustment with elevated bilirubin levels | (82,83) |
| Etoposide | In pre-existing liver disease: Mild to moderate impairment: no pharmacokinetic effect, Severe impairment: myelosuppression, mucositis. Decreased albumin increases unbound drug concentration and increases haematological toxic effects Veno-occlusive disease in higher doses Case reports of severe hepatocellular damage at standard doses | Common in bone-marrow transplant-tation (10–25%) Rare | Severe and life-threatening Severe | Unclear (increase renal crearance might compensate) | (84-86) |
| Gemcitabine | Increased aminotransferases Increased bilirubin Case reports of fatal cholestatic hepatotoxicity | Veri common (up To 60%) Rare | Generally transient and reversible Deterioration of Liver function Can be fatal | No dose adjustment Reduce dose (20%) and increased if tolerated | (60-62) |
| Imatinib | Increased aminotransferases or bilirubin Liver necrosis of failure | 5–8% Rare | 2–8% grade 3–4 Case reports of Fatalities | Stop treatment if hepatotoxicity develops, should probably not Rechallenge | (64-66) |
| Sorafenib | In pre-existing liver disease: clearance does not differ between patient cohorts | Dose adjustment with elevated bilirubin | (67) |
Drugs can be devided into dose-dependent or predictable hepatotoxins, and dose-independent or unpredictable hepatotoxins. Dose-dependent hepato-toxins generally require metabolic activation to a toxic metabolite and interfere with subcellular orga-nelles and biochemical processes at key sites such as mitochondria or canalicular bile secretion.8,36 Liver injury produced by dose-dependent hepatotoxins usually occurs after a short latent period (hours), and is characterized by zonal necrosis or microvesi-cular steatosis (i.e. aflatoxins).37
Most clinically overt adverse hepatic events associated with drugs are unpredictable. These events can be divided into apparent hypersensitivi-ty reactions associated with fever, rash, eosino-philia, and a rapid response upon rechallenge, versus presumed metabolic unpredictable (idiosyncratic) reactions.13
5-Fluorouracil (5-FU)5-FU is the base of most combination therapies of gastrointestinal tumors. It is an uracil analogue and antimetabolite that is metabolized mainly in the liver by dihydropyrimidindehydrogenase (DPD). In case of DPD-deficiency myelotoxicity and diarrhea are the most common toxic effects, followed by mu-cositis, hand-foot-syndrome and neurotoxicity.38 5-FU has been associated with hepatic steatosis on histological and radiological findings, but has not been shown to affect morbidity or mortality.39–41 5-FU is fairly safe to use in patients with liver dysfunction, although regular monitoring of liver tests is advised.42
Cαpecitαbine is an oral prodrug that is converted to fluorouracil in three enzymatic steps via carboxylesterase to 5’-Desoxy-5-Fluorocytidin (5’-DFCR), cytidin-deaminase to 5’-Desoxy-5-Fluorouri-din (5’-DFUR) and by thymidine phosphorylase (TP) to the active component 5-FU. TP is an enzyme which is present at higher levels in some tumors and the liver than in other healthy tissue.43 Hyperbilirubi-naemia is common and usually reversible.44,45 It might be related to haemolysis.46
OxaliplatinOxaliplatin is a platinum analogue which ist given in combination with 5-FU. It belongs to the alkylating chemotherapy agents which, like Cis-or Carboplatin, result in the formation of crosslinks within and between DNA strands. Oxaliplatin is mainly excreted through the kidneys and does not undergo cytochrome P450 metabolism within the liver. Hence it is tolerated by patients with liver dys-function.47 Nevertheless, studies of patients undergoing liver metastasectomy after neoadjuvant oxaliplatin-based treatment have shown histological evidence of liver damage.48–51 Neoadjuvant chemotherapy with 5-FU and oxaliplatin-based regimes has been associated with vascular changes in heal-thy liver parenchyma, more than steatosis. Frequency of oxaliplatin-associated sinusoidal obstruction or dilatation syndrome varies in reports from 20–80%,49–51 a condition which might increase morbidity but not mortality after liver metastec-tomy. Current recommendations are to limit the number of preoperative cycles if possible.35
IrinotecanIrinotecan (CPT-II) is the other main combination partner of 5-FU in advanced colorectal carcinoma therapy. It is a camptothecin analogue, and is a prodrug which requires bioactivation in the liver by carboxyesterase to form the active and lipophilic metabolite SN-38. SN-38 acts as a topoisomerase I poison and is approximately 100–1000-fold more cytotoxic than the parent drug.52 Topoisomerases are enzymes which are responsible for the maintenance of DNA-topology during the S-phase. Irinote-can and its active metabolite SN-38 form a complex with topoisomerase I and inhibit DNA-replication. A similar substance is topotecan. Etoposid inhibits DNA-Topoisomerase II.53
Irinotecan is subject to extensive metabolic conversion by various enzyme systems, including esterases to form SN-38, UGT1A1 mediating glucuronidation of SN-38, as well as CYP3A4, which forms several pharmacologically inactive oxidation products. Elimination routes of irinotecan also depend on the presence of drug-transporting proteins, notably P-glycoprotein and canalicular multispecific organic anion transporter, present on the bile cana-licular membrane. The various processes mediating drug elimination, either through metabolic breakdown or excretion, likely impact substantially on inter-individual variability.54
Toxic levels of irinotecan can accumulate from standard doses if glucuronidation is reduced, as occurs in Gilbert’s-Meulengracht syndrome.35 Elevated bilirubin and AP levels are associated with decreased irinotecan clearance.55
Irinotecan is associated with hepatic steatosis and steatohepatitis in up to 50% of liver-resection samples.49–56 Steatohepatitis has been associated with increased morbidity, and increased mortality in some series, after liver resection.51
It is generally more common to use oxaliplatin-based regimens than those that are irinotecan-based for neoadjuvant treatment before liver resection.35 Irinotecan can also cause mild elevations of serum transaminases and bilirubin in up to 25% of pa-tients.57
GemcitabineGemcitabine (2’,2’-Difluordesoxycytidin) is a pyri-midine/deoxycytidine analogue which acts as anti-metabolite. It is a pro-drug that, once transported into the cell, undergoes intracellular phosphoryla-tion and activation by deoxycytidine kinase. Both gemcitabine diphosphate and gemcitabine triphos-phate inhibit processes required for DNA synthesis. Incorporation of gemcitabine triphosphate (dFdC-TP) into DNA is most likely the major mechanism by which gemcitabine causes cell death. After incorporation of the gemcitabine nucleotide on the end of the elongating DNA strand, one deoxynucleotide is added and DNA polymerases are unable to proceed, a process called masked termination.58
Active gemcitabine triphosphate is degraded by cytidine deaminase in liver, blood and kidney to form an inactive metabolite: 2’,2’-difluorodeoxyuri-dine (dFdU).59 It is eliminated by the kidney.
Administration of gemcitabine is associated with a mild rise in transaminases in up to 60% of patients, and in serum bilirubin in a small proportion of patients. These changes are usually transient, but are very rarely associated with severe hepatotoxici-ty. 60,61 Monitoring of liver tests in patients who are receiving gemcitabine is recommended, the expected mild rise in transaminases is usually of no clinical consequence.35 In case of slightly elevated transami-nases, gemcitabine is generally well tolerated without increased toxicity. However, in patients with primarily elevated bilirubin, significant deterioration in liver function under therapy has been described. Patients with elevated creatinine levels had significant toxicity even at reduced doses.62
DacarbacineDacarbazine is an alkylating agent and crosslinks DNA during all phases of the cell cycle, resulting in disruption of DNA function, cell cycle arrest, and apoptosis. It is used in the therapy of advanced neuroendocrine tumors, malignant melanoma, hodgkins lymphoma and sarcoma. Common side-effects include nausea and vomiting and bone-marrow suppression. There are case reports which indicate that next to elevation of liver enzymes, there might be hepatic veno-oclusive disease associated with the administration of Dacarbazine.63
ImatinibImatinib is an oral small-molecule tyrosine kina-se inhibitor, effective to inhibit the BCR-ABL kinase in chronic myeloid leukaemia and the KIT tyrosine kinase and platelet-derived growth factor receptor in gastrointestinal stromal tumors (GISTs). Cases of severe liver injury are rare. In the two large multicentre studies with 147 and 946 patients treated for GIST, abnormal liver function tests were observed in 5.4% of treated patients (including grade 3–4 toxicity in 2.7%), and fatal liver disease occurred in 3%.64,65 In chronic myeloic leukemia imatinib was found to cause grade 2 or higher elevations in serum aminotransferases in 8% of the treated patients and grade 3–4 elevations in 1–5%. Grade 3–4 elevations in serum bilirubin levels were reported in 0.4–3.5% of the patients.66
SorafenibSorafenib is an orally available multi-kinase inhibitor, which has shown effectivity against hepatoce-llular carcinoma and renal-cell carcinoma. It is mainly metabolized in the liver through both, an oxidative pathway mediated by cytochrome P450 and UGT 1A9-mediated glucuronidation; excretion is mostly faecally.34 A phase I study of sorafenib in patients with liver dysfunction who were devided into groups on the basis of bilirubin levels, albumin levels, and renal function, showed that although drug clearance did not differ between groups, dose-limiting toxic effects occurred with increasing biliru-bin levels and decreased albumin.67
Cetuximab and panitumumabThe epidermal growth factor receptor (EGFR) is a member of the ErbB family of closely related tyrosine kinase receptors: EGFR (ErbB-l/HER-1), ErbB-2 (HER-2/neu), ErbB-3 (HER-3), and ErbB-4 (HER-4). Cetuximab and panitumumab are monoclonal antibodies that block the ligand binding site of the EGFR, thus inhibiting intracellular signaling. Cetuximab is a chimeric human-mouse antibody, while panitumu-mab is a fully humanized monoclonal anti-EGFR an-tibody. Common side effects of these antibodies include acneiform rash, diarrhea, and hypomagnesemia and hypersensitivity reactions that can be particularly severe with the chimeric antibody.68 Recent retrospective analyses from several large trials show that patients with tumors bearing the KRAS mutation do not respond to either cetuximab-or panitumumab-ba-sed therapy.69–71 These antibodies are generally used in combination with 5-FU based therapy. However there are positive results for monotherapy in advanced disease. In a case of chronic cholestasis and hy-perbilirubinemia caused by advanced liver involvement which prohibits second-line treatment, combined antibody treatment with cetuximab and be-vacizumab (an antibody against vascular endothelial growth factor) was given and well-tolerated. Clinical performance status as well as laboratory parameters improved rapidly and combined chemotherapy could be administered.72
BevacizumabBevacizumab is a humanized antibody targeting vascular endothelial growth factor A (VEGF-A).68 It has been approved in first-and second-line treat-ment of metastatic colorectal cancer in combination with 5-FU and in lung cancer in combination with carboplatin and paclitaxel. It has also shown benefit in metastatic breast cancer when combined with pa-clitaxel.73 The relatively well-known side effects include hypertension, gastrointestinal perforation, arterial thromboembolic events and postoperative bleeding or wound-healing complications. Treatment with this antibody is safe, even with elevated liver values. Bevacizumab has no impact on hepatic stea-tosis and fibrosis.74 As for sinusoidal obstructions, results are contradictory. One report states that be-vacizumab protects against the sinusoidal obstruction syndrome,74 whilst there are case reports of hepatic injury including veno-occlusive disease75 and singular lesions with necrotic liver parenchyma with inflammatory reaction.73 The reported side-effects however resolved after cessation of the thera-py.73 Even though these reactions are rare and unusual, one should be aware that bevacizumab might cause portal hypertension with elevation of transaminases and bilirubin.
Risks of Neoadjuvant ChemotherapyThe changes (steatosis, steatohepatitis, sinusoidal injury) described above may be responsible for complications after surgery when neoadjuvant chemotherapy is performed in patients with advanced cancer. The preoperative chemotherapy however has several advantages: 1) it downsizes tumors, 2) it increases curative resection rates and 3) it converts some patients from having unresectable to resectable disease. This kind of therapy has been however associated with an increased risk of perioperative morbidity or 90 days mortality.76,77 In general, there is no increase in complications even in the presence of severe (> 30%) steatosis, when steatohepatitis (stea-tosis with inflammation) is excluded.78
Hepatic steatosis, a mild manifestation of non-alcoholic fatty liver disease (NAFLD), may occur after treatment with 5-FU.41 Steatohepatitis is for instance associated with irinotecan therapy. Hepatic sinusoidal obstruction can occur with oxaliplatin treatment and appears to increase in severity with prolonged treatment (> 6 cycles). It does however not appear to be associated with an increased risk of perioperative death.41,78 Bevacizumab can be used safely when discontinued > 5 weeks before liver re-section.79
Future AspectsCancer continues to be one of the major causes of mortality throughout the world. Modern combination chemotherapy allows for a longer time-to-progression (TTP) and longer overall survival (OS) even in advanced disease - resulting in a prolonged treatment. Longer treatment times lead to increased toxic effects, especially in an aging patient collective. Whilst the maximal tolerable dose (MTD) might be given at the beginning of a therapy and particularly in the neoadjuvant and adjuvant treatment settings, it might not be possible to continue this treatment over a longer period, especially in palliative treatment.
Therefore, we should ask ourselves, what would be the minimal effective dose (MED) to treat our long-term patients, and when to change. Whilst prognostic biomarkers help to give us information about the therapeutic outcome of a specific disease, predictive biomarkers might help us to anticipate the effect of a specific therapy for this disease whilst keeping the side effects tolerable.
Accounting for further individual parameters of the patients such as age, gender and disease stage, matched to the expected side effects under a given dose, the efficacy of a treatment as well as the quality of life of our tumor patients, shall be maintained as good as possible.



