



Introduction and aim. Approximately 650,000 people in Brazil have chronic hepatitis C virus (HCV) infection. We evaluated the safety and efficacy of ombitasvir (OBV)/paritaprevir (PTV)/ritonavir (r) plus dasabuvir (DSV) with/without ribavirin (RBV) in an open-label multicenter phase 3b trial in treatment-naive or interferon (IFN) treatment-experienced Brazilian patients with advanced hepatic fibrosis (METAVIR F3/4) and HCV genotype (GT) 1 infection.
Material and methods. All patients received coformulated OBV/ PTV/r daily + DSV twice daily (3-DAA). GTIa-infected patients received 3-DAA plus RBV for 12 weeks, except for prior pegIFN/ RBV nonresponders with cirrhosis who were treated for 24 weeks. GTIb-infected patients received 3-DAA alone (F3) or in combination with RBV (F4) for 12 weeks. The primary endpoint was sustained virologic response (HCV RNA < 15 IU/mL) at post-treatment Week 12 (SVR12).
Results. The study enrolled 222 patients, 214 achieved an SVR12 (96.4%; 95% CI, 93.1-98.2%), one GT1a-infected patient experienced virologic breakthrough, six (5 GT1a) relapsed, and one was lost to follow-up. SVR12 was achieved in 111/ 112 (99.1%) GT1b-infected patients, including 42/43 (97.7%) noncirrhotic, and 69/69 (100%) cirrhotic patients; and in 103/110 (93.6%) GT1a-infected patients, including 44/46 (95.7%) noncirrhotic and 59/64 (92.2%) cirrhotic patients. Overall there was a low rate of serious adverse events (n = 6, 2.7%). One patient experienced a treatment-related serious adverse event and one patient discontinued treatment because of an adverse event.
Discussion. The results confirm that the 3-DAA regimen with/without RBV is well tolerated and had a favorable safety profile and is efficacious in GT1-infected patients with advanced fibrosis (METAVIR F3/4).
The seroprevalence of chronic hepatitis C virus (HCV) infection in Brazil is estimated to be approximately 1.38%, translating to currently 650,000 people who require treatment, most of whom are not diagnosed.1–4 In 2017, Brazil’s universal healthcare system updated its Guideline and now provides free treatment for citizens with chronic hepatitis C including individuals with HIV coinfection, renal insufficiency, idiopathic thrombocytopenic purpura, cryoglobulinemia, hematologic malignancies, solid organ transplant recipients, severe extra-hepatic manifestations, and moderate hepatic fibrosis (METAVIR stages greater than F2 by biopsy or non-invasive methodology).5 HCV genotype (GT) 1 is the most common across all regions of Brazil;6 thus, a regimen must have good activity against GT1a and GT1b to be suitable for use in this setting.
Across many regions of the world the current standard of care for patients with chronic hepatitis C involves treatment with combinations of orally administered direct-acting antiviral agents (DAAs).7–9 The combination of ombitasvir (OBV), ritonavir-enhanced paritaprevir (PTV/r; paritaprevir developed by AbbVie and Enanta), and dasabuvir (DSV), with or without weight-based ribavirin (RBV), has demonstrated high rates of sustained virologic response at post-treatment Week 12 (SVR12) in GTl-infected patients without cirrhosis or with compensated cirrhosis,10–15 and is one of several regimens recommended for this subset of patients in current treatment guidelines.7–9
The objective of this Phase 3b study was to evaluate the efficacy and safety of 12 or 24 weeks of treatment with OBV/PTV/r plus DSV (3-DAA) with or without RBV in treatment-naive or interferon (IFN) treatment-experienced, GT1-infected Brazilian patients with advanced hepatic fibrosis (bridging fibrosis or compensated cirrhosis, METAVIR F3-F4).
Material and MethodsTOPAZ-III (M14-225, clinicaltrials.gov identifier NCT02442271) was a Phase 3b, open-label, multicenter trial that was conducted at 16 sites in Brazil. The study conformed to the Declaration of Helsinki and adhered to International Conference on Harmonization (ICH) guidelines. The protocol and all amendments were approved by independent ethics committees or institutional review boards at each study site. All patients provided written informed consent prior to undergoing any study procedures.
PatientsPatients eligible for the trial were adults ≥ 18 years of age with HCV genotype 1 infection, a serum HCV RNA level > 1000 IU/mL at the time of screening and advanced hepatic fibrosis. Advanced fibrosis was documented by the results of a liver biopsy performed within 24 months prior to receipt of the first dose of study drug (METAVIR F3, Ishak 4, or equivalent); a liver biopsy showing cirrhosis (METAVIR F3/4 or F4, Ishak 5 or 6, or equivalent) at any time prior to receipt of the first dose of study drug; a FibroScan® result of ≥ 9.6 kPa obtained within 6 months prior to the first dose of study drug; or a FibroTest® result of ≥ 0.59 obtained during the screening period. Patients with cirrhosis could be included if they had compensated liver disease (Child-Pugh A) with no prior history of hepatic decompensation. Both treatment-naive and IFN-experienced patients were eligible.
Patients with cirrhosis were required to have ultrasound, computed tomography or magnetic resonance imaging within 3 months prior to screening in order to exclude hepatocellular carcinoma.
Patients were excluded if they had a positive screening test result for human immunodeficiency virus or hepatitis B surface antigen. Patients were also ineligible if they had a creatinine clearance < 30 mL/min, albumin < 2.8 g/dL, hemoglobin < 10 g/dL, platelet count < 25 × 109/L or a total bilirubin level > 3 mg/dL. Patients who had previously received direct acting antiviral agents were ineligible for the trial.
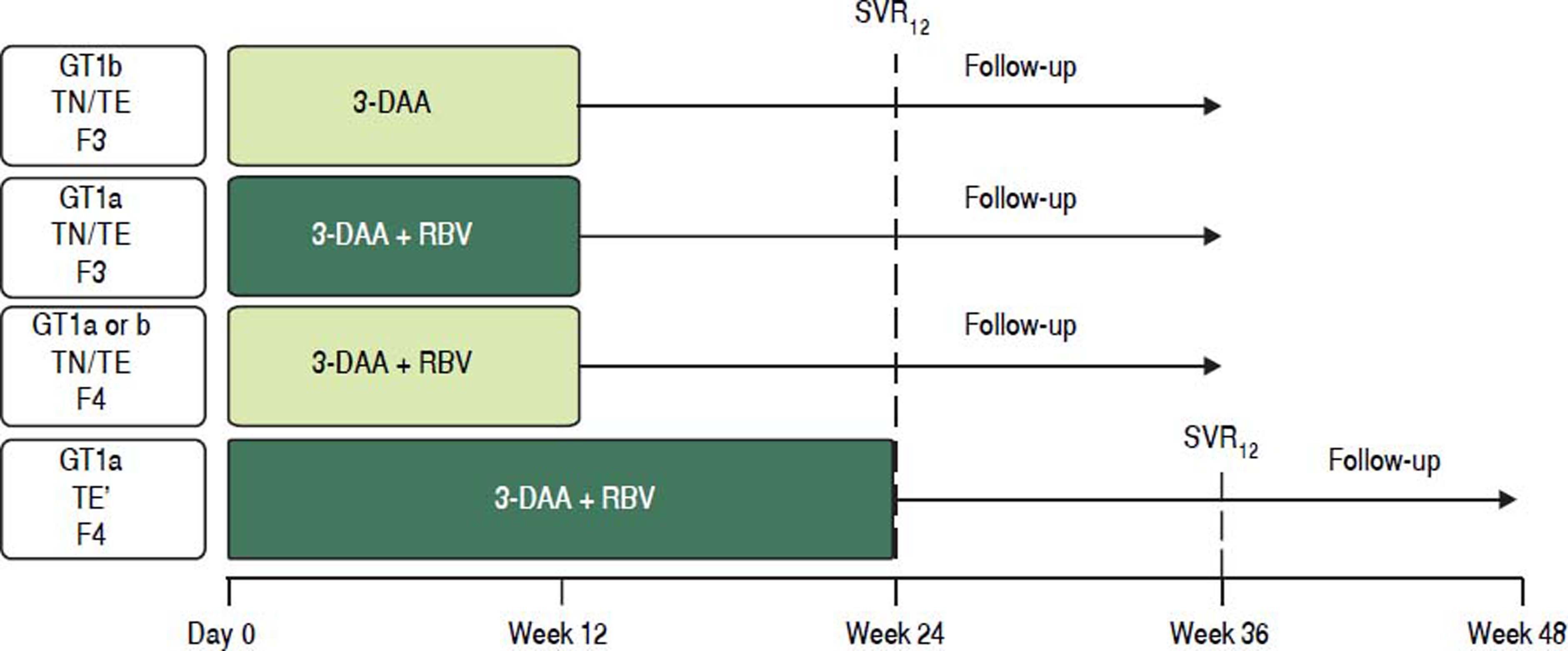
TreatmentAll patients received oral co-formulated OBV/PTV/r (25/150/100 mg) once daily and DSV (250 mg twice daily) with or without RBV (1,000 mg daily for body weight < 75 kg or 1,200 mg daily for body weight ≥ 75 kg) for 12 or 24 weeks according to HCV GT1 subtype (1a or 1b) and METAVIR fibrosis stage (Figure 1). All noncirrhotic patients (GT1a- and GT1b-infected) and cirrhotic GT1b-infected patients were assigned to 12 weeks of treatment regardless of treatment history (naive or IFN-experienced). Among GT1a-infected patients with cirrhosis, those who were treatment naive, or who had experienced a prior partial virologic response, breakthrough or relapse, or who were intolerant to previous treatment with IFN-based therapy were assigned to 12 weeks of treatment; whereas, GT1a-infected patients with cirrhosis who had experienced a prior nonresponse or null response to previous treatment with IFN-based therapy were assigned to 24 weeks of treatment. All GT1a-infected patients and GT1b-infected patients with cirrhosis received RBV. Noncirrhotic patients with GT1b infection received the regimen without RBV.
. TN: treatment-naive. TE: treatment-experienced (IFN/RBV or pegIFN/RBV experienced patients).")
Study Design. * Treatment experienced patients with cirrhosis and GT1a infection and a prior null response or nonresponse were assigned to 24 weeks of treatment Al other patients with cirrhosis were assigned to 12 weeks of treatment 3-DAA, ombitasvir/paritaprevir/ritonavir 25/150/100 mg once daily + dasabuvir 250 mg twice daily ± ribavirin (weight-based). TN: treatment-naive. TE: treatment-experienced (IFN/RBV or pegIFN/RBV experienced patients).
All patients who received at least one dose of study drug were followed up for 24 weeks after completion or premature discontinuation of study drug.
Efficacy and safety assessmentsThe primary efficacy endpoint was sustained virologic response (the proportion of patients with HCV RNA below the lower limit of quantification [LLOQ]) at 12 weeks (SVR12) after the final dose of study drug. HCV RNA levels in plasma were determined using the COBAS® AmpliPrep/COBAS® Taqman® HCV Test, v. 2.0 (LLOQ = 15 IU/mL). Secondary efficacy endpoints were SVR12 rates by fibrosis stage, prior treatment experience (naive, or previous IFN-based regimen), and IFN eligibility (ineligible, intolerant, eligible).
Treatment-emergent adverse events (TEAEs) were monitored from initiation of treatment until 30-days after discontinuation of study drugs.
Resistance analysesFor any patient who experienced virologic failure, HCV regions encoding NS3/4A, NS5A, and NS5B were amplified by PCR and the resulting PCR products were sequenced by population sequencing (approximately 15% detection threshold) from the baseline sample and the first sample obtained after virologic failure that had an HCV RNA level ≥ 1000 IU/mL. The resulting sequences were analyzed for the presence of pre-existing baseline polymorphisms and treatment-emergent amino acid substitutions.
Data analysesGiven a planned enrollment of 220 patients and an estimated SVR12 rate of 95%, the 2-sided 95% confidence interval (CI) calculated by Wilson’s score method will be 91.3% to 97.2%. No adjustments were made for dropouts. Analyses were performed on the intent-to-treat (ITT) population, which consisted of all patients who received at least one dose of study drug. Patients with missing SVR12 results were considered to be treatment failures.
Safety data were analyzed using descriptive statistics.
ResultsThe study enrolled 222 individuals (ITT population), including 59.9% (n = 133) with cirrhosis (48.1% [64/133] and 51.9% [69/133] with GT1a and GT1b infection, respectively) and 40.1% (n = 89) noncirrhotic (F3) patients (51.7% [46/89] and 48.3% [43/89] with GT1a and GT1b infection, respectively) across 16 study centers in Brazil. The first patient was enrolled on 27 April 2015 and the last patient completed follow-up on 26 September 2016.
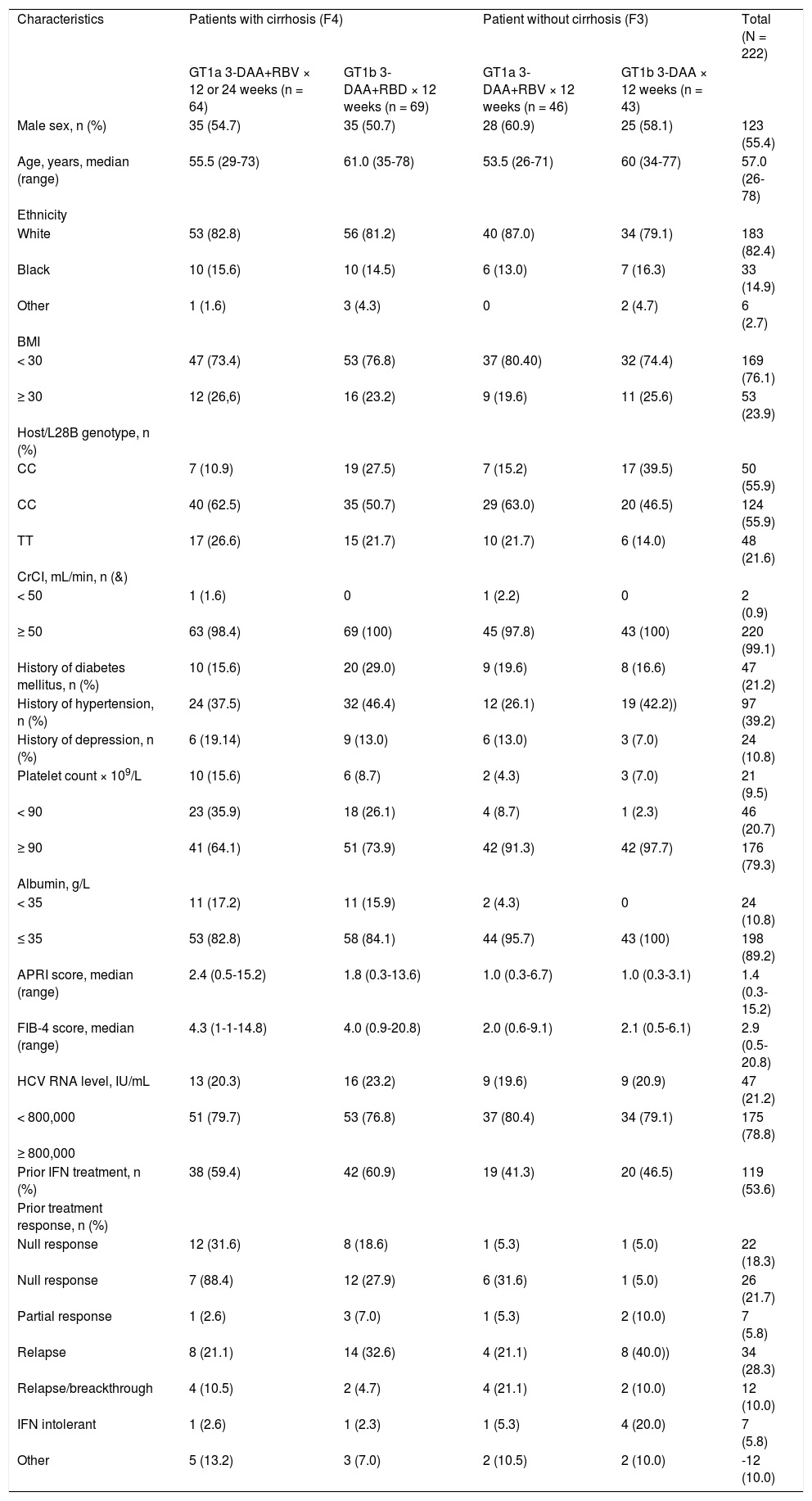
The majority of patients were male (55.4%), white (82.4%), aged < 65 years (77.0%), and had received prior IFN-based therapy (54.1%) (Table 1). Overall, the median age was 57 years (range 26-78), and the median BMI was 26.5 kg/m2 (range 17–51).
Baseline demographic and clinical characteristics.
| Characteristics | Patients with cirrhosis (F4) | Patient without cirrhosis (F3) | Total (N = 222) | ||
| GT1a 3-DAA+RBV × 12 or 24 weeks (n = 64) | GT1b 3-DAA+RBD × 12 weeks (n = 69) | GT1a 3-DAA+RBV × 12 weeks (n = 46) | GT1b 3-DAA × 12 weeks (n = 43) | ||
| Male sex, n (%) | 35 (54.7) | 35 (50.7) | 28 (60.9) | 25 (58.1) | 123 (55.4) |
| Age, years, median (range) | 55.5 (29-73) | 61.0 (35-78) | 53.5 (26-71) | 60 (34-77) | 57.0 (26-78) |
| Ethnicity | |||||
| White | 53 (82.8) | 56 (81.2) | 40 (87.0) | 34 (79.1) | 183 (82.4) |
| Black | 10 (15.6) | 10 (14.5) | 6 (13.0) | 7 (16.3) | 33 (14.9) |
| Other | 1 (1.6) | 3 (4.3) | 0 | 2 (4.7) | 6 (2.7) |
| BMI | |||||
| < 30 | 47 (73.4) | 53 (76.8) | 37 (80.40) | 32 (74.4) | 169 (76.1) |
| ≥ 30 | 12 (26,6) | 16 (23.2) | 9 (19.6) | 11 (25.6) | 53 (23.9) |
| Host/L28B genotype, n (%) | |||||
| CC | 7 (10.9) | 19 (27.5) | 7 (15.2) | 17 (39.5) | 50 (55.9) |
| CC | 40 (62.5) | 35 (50.7) | 29 (63.0) | 20 (46.5) | 124 (55.9) |
| TT | 17 (26.6) | 15 (21.7) | 10 (21.7) | 6 (14.0) | 48 (21.6) |
| CrCI, mL/min, n (&) | |||||
| < 50 | 1 (1.6) | 0 | 1 (2.2) | 0 | 2 (0.9) |
| ≥ 50 | 63 (98.4) | 69 (100) | 45 (97.8) | 43 (100) | 220 (99.1) |
| History of diabetes mellitus, n (%) | 10 (15.6) | 20 (29.0) | 9 (19.6) | 8 (16.6) | 47 (21.2) |
| History of hypertension, n (%) | 24 (37.5) | 32 (46.4) | 12 (26.1) | 19 (42.2)) | 97 (39.2) |
| History of depression, n (%) | 6 (19.14) | 9 (13.0) | 6 (13.0) | 3 (7.0) | 24 (10.8) |
| Platelet count × 109/L | 10 (15.6) | 6 (8.7) | 2 (4.3) | 3 (7.0) | 21 (9.5) |
| < 90 | 23 (35.9) | 18 (26.1) | 4 (8.7) | 1 (2.3) | 46 (20.7) |
| ≥ 90 | 41 (64.1) | 51 (73.9) | 42 (91.3) | 42 (97.7) | 176 (79.3) |
| Albumin, g/L | |||||
| < 35 | 11 (17.2) | 11 (15.9) | 2 (4.3) | 0 | 24 (10.8) |
| ≤ 35 | 53 (82.8) | 58 (84.1) | 44 (95.7) | 43 (100) | 198 (89.2) |
| APRI score, median (range) | 2.4 (0.5-15.2) | 1.8 (0.3-13.6) | 1.0 (0.3-6.7) | 1.0 (0.3-3.1) | 1.4 (0.3-15.2) |
| FIB-4 score, median (range) | 4.3 (1-1-14.8) | 4.0 (0.9-20.8) | 2.0 (0.6-9.1) | 2.1 (0.5-6.1) | 2.9 (0.5-20.8) |
| HCV RNA level, IU/mL | 13 (20.3) | 16 (23.2) | 9 (19.6) | 9 (20.9) | 47 (21.2) |
| < 800,000 | 51 (79.7) | 53 (76.8) | 37 (80.4) | 34 (79.1) | 175 (78.8) |
| ≥ 800,000 | |||||
| Prior IFN treatment, n (%) | 38 (59.4) | 42 (60.9) | 19 (41.3) | 20 (46.5) | 119 (53.6) |
| Prior treatment response, n (%) | |||||
| Null response | 12 (31.6) | 8 (18.6) | 1 (5.3) | 1 (5.0) | 22 (18.3) |
| Null response | 7 (88.4) | 12 (27.9) | 6 (31.6) | 1 (5.0) | 26 (21.7) |
| Partial response | 1 (2.6) | 3 (7.0) | 1 (5.3) | 2 (10.0) | 7 (5.8) |
| Relapse | 8 (21.1) | 14 (32.6) | 4 (21.1) | 8 (40.0)) | 34 (28.3) |
| Relapse/breackthrough | 4 (10.5) | 2 (4.7) | 4 (21.1) | 2 (10.0) | 12 (10.0) |
| IFN intolerant | 1 (2.6) | 1 (2.3) | 1 (5.3) | 4 (20.0) | 7 (5.8) |
| Other | 5 (13.2) | 3 (7.0) | 2 (10.5) | 2 (10.0) | -12 (10.0) |
One cirrhotic patient who was assigned to 12 weeks of treatment with 3-DAA plus RBV prematurely discontinued study drug due to a TEAE; thus, 99.5% of patients (221/222) completed treatment.
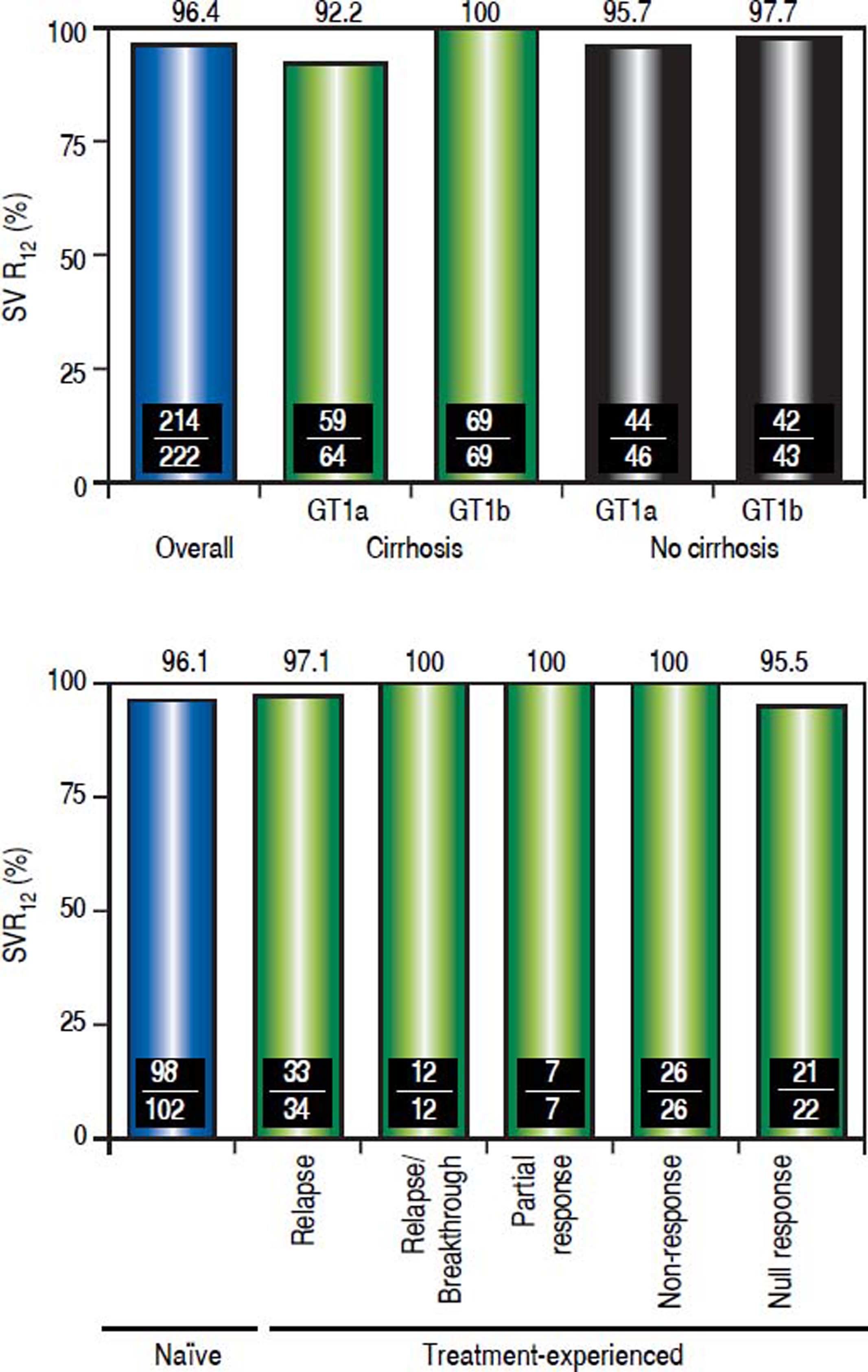
EfficacyThe SVR12 rate was 96.4% (214/222; 95% CI, 93.198.2%) (Figure 2A). Among the 8 patients who did not achieve SVR12, 1 patient experienced on-treatment virologic failure, 6 patients experienced post-treatment relapse and 1 GT1a-infected patient with undetectable HCV RNA at post-treatment Week 4 was subsequently lost to follow-up and had a missing HCV RNA value during the SVR12 assessment window. The SVR24 rate was 95.5% (212/222; 95% CI, 91.9-97.5%). The 2 additional patients who did not achieve SVR24 had missing HCV RNA values during the SVR24 assessment window (the patient with a missing HCV RNA value during the SVR12 assessment window also had a missing HCV RNA value during the SVR24 assessment window). No late relapses were documented between follow-up Week 12 and follow-up Week 24, although two patients were lost to follow-up.

Sustained virologic response rates overall, and by HCV subtype, hepatic fibrosis status and previous response to pegIFN/RBV combination therapy. Definitions of responses to prior treatment with pegIFN/RBV: Relapse, undetectable HCV RNA at the end of treatment, but was detected during follow-up; Relapse/breakthrough, ≥ 1 documented result of undetectable HCV RNA during treatment; Nonresponse, no documented undetectable HCV RNA results during treatment, or HCV RNA was detected at the end of treatment with insufficient data available to categorize the response as a relapse, breakthrough, partial response or null response; Partial response, ≥ 2-log IU/mL decrease in HCV RNA by Week 12; Null response, < 1-log IU/mL reduction in HCV RNA by Week.
The SVR12 rate was 99.1% (111/112) in patients with GT1b infection, and 93.6% (103/110) in patients with GT1a infection. SVR12 was achieved in 96.2% (128/133) of cirrhotic patients and 96.6% (86/89) of noncirrhotic patients. Overall, 96.1% (98/102) of treatment-naive patients, and 96.7% (116/120) of patients who had received prior IFN-based treatment achieved an SVR12 (p = not significant, Figure 2B). Specifically, SVR12 was achieved by 97.1% in previous relapsers, 100% in patients with previous relapse/breakthrough, partial response or nonresponse, and 95.5% in patients with a prior null response to IFN/RBV or pegIFN/RBV combination therapy.
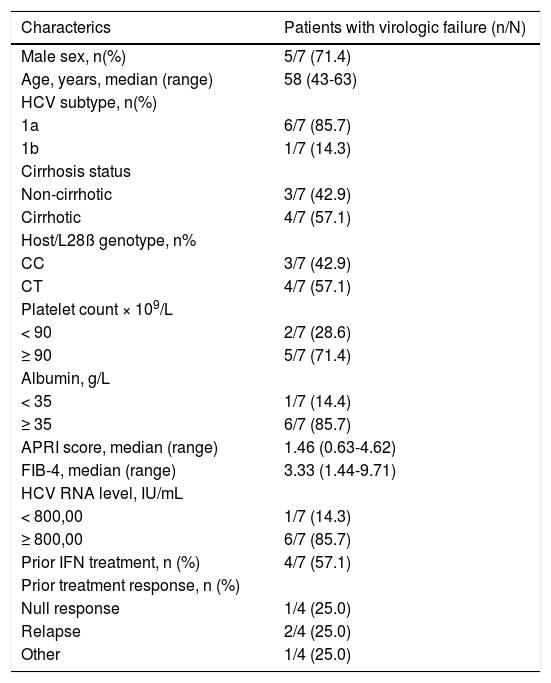
The baseline characteristics of the patients who experienced virologic failure are presented in table 2.
Baseline characterictics of patients who experienced virologic failure.
| Characterics | Patients with virologic failure (n/N) |
|---|---|
| Male sex, n(%) | 5/7 (71.4) |
| Age, years, median (range) | 58 (43-63) |
| HCV subtype, n(%) | |
| 1a | 6/7 (85.7) |
| 1b | 1/7 (14.3) |
| Cirrhosis status | |
| Non-cirrhotic | 3/7 (42.9) |
| Cirrhotic | 4/7 (57.1) |
| Host/L28ß genotype, n% | |
| CC | 3/7 (42.9) |
| CT | 4/7 (57.1) |
| Platelet count × 109/L | |
| < 90 | 2/7 (28.6) |
| ≥ 90 | 5/7 (71.4) |
| Albumin, g/L | |
| < 35 | 1/7 (14.4) |
| ≥ 35 | 6/7 (85.7) |
| APRI score, median (range) | 1.46 (0.63-4.62) |
| FIB-4, median (range) | 3.33 (1.44-9.71) |
| HCV RNA level, IU/mL | |
| < 800,00 | 1/7 (14.3) |
| ≥ 800,00 | 6/7 (85.7) |
| Prior IFN treatment, n (%) | 4/7 (57.1) |
| Prior treatment response, n (%) | |
| Null response | 1/4 (25.0) |
| Relapse | 2/4 (25.0) |
| Other | 1/4 (25.0) |
Seven of the 8 patients who did not achieve SVR12 experienced virologic failure. Six individuals (5 infected with GT1a, 1 with GT1b) experienced relapse during follow-up, and 1 individual (GT1a-infected) experienced on-treatment virologic failure (breakthrough at Week 12).
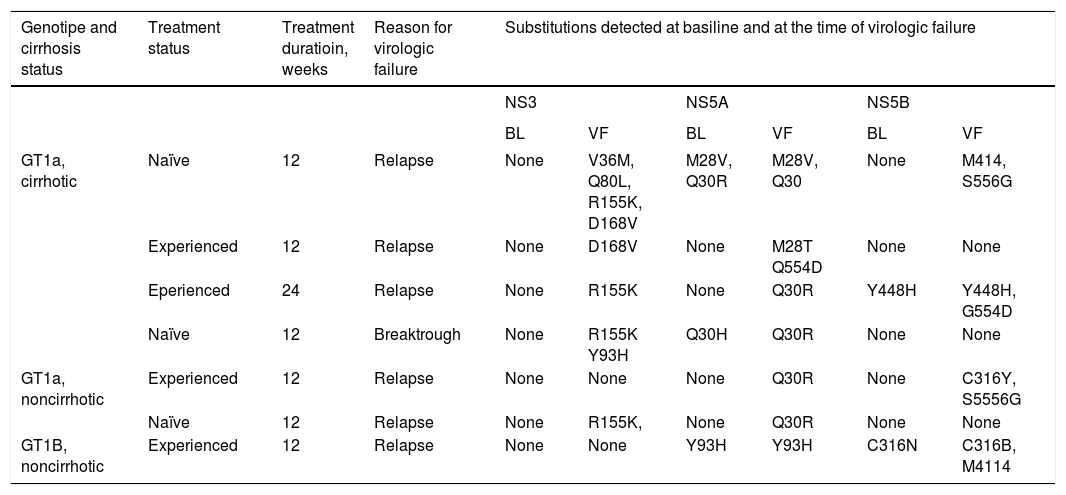
Substitutions at resistance-associated amino acid positions in NS3, NS5A, or NS5B in patients who experienced virologic failure are shown in table 3. NS3 substitutions were not detected at baseline in any patients, but were detected at the time of virologic failure in 5 patients. NS5A substitutions were detected in 3 patients at baseline and in all 7 patients at the time of virologic failure. NS5B substitutions were detected in 2 patients at baseline and in 4 patients at the time of virologic failure. The most common treatment-emergent substitutions were R155K and D168V in NS3, M28T and Q30R in NS5A, and C316Y, M414I, G554D, and S556G in NS5B.
Patients with virologic failure: NS, NS5A, and NS5B substitutions at baseline at the time of virologic failure.
| Genotipe and cirrhosis status | Treatment status | Treatment duratioin, weeks | Reason for virologic failure | Substitutions detected at basiline and at the time of virologic failure | |||||
|---|---|---|---|---|---|---|---|---|---|
| NS3 | NS5A | NS5B | |||||||
| BL | VF | BL | VF | BL | VF | ||||
| GT1a, cirrhotic | Naïve | 12 | Relapse | None | V36M, Q80L, R155K, D168V | M28V, Q30R | M28V, Q30 | None | M414, S556G |
| Experienced | 12 | Relapse | None | D168V | None | M28T Q554D | None | None | |
| Eperienced | 24 | Relapse | None | R155K | None | Q30R | Y448H | Y448H, G554D | |
| Naïve | 12 | Breaktrough | None | R155K Y93H | Q30H | Q30R | None | None | |
| GT1a, noncirrhotic | Experienced | 12 | Relapse | None | None | None | Q30R | None | C316Y, S5556G |
| Naïve | 12 | Relapse | None | R155K, | None | Q30R | None | None | |
| GT1B, noncirrhotic | Experienced | 12 | Relapse | None | None | Y93H | Y93H | C316N | C316B, M4114 |
Amino acid positions included in the analysis of virus from patients with GT1a infection: 36, 43, 55, 56, 80, 155, 156, 168 in NS3; 24, 28, 29, 30, 31, 32, 58, 62, 92, 93 in NS5A; 316, 414, 446, 448, 451, 553, 554, 555, 556, 558, 559, 561 in NS5B. Amino acid positions included in analysis of virus from patients with GT1b infection: 55, 56, 155, 156, 168 in NS3; 24, 28, 29, 30, 31, 32, 58, 62, 92, 93 in NS5A; 316, 368, 411, 445, 448, 553, 556, 558, 559 in NS5B.
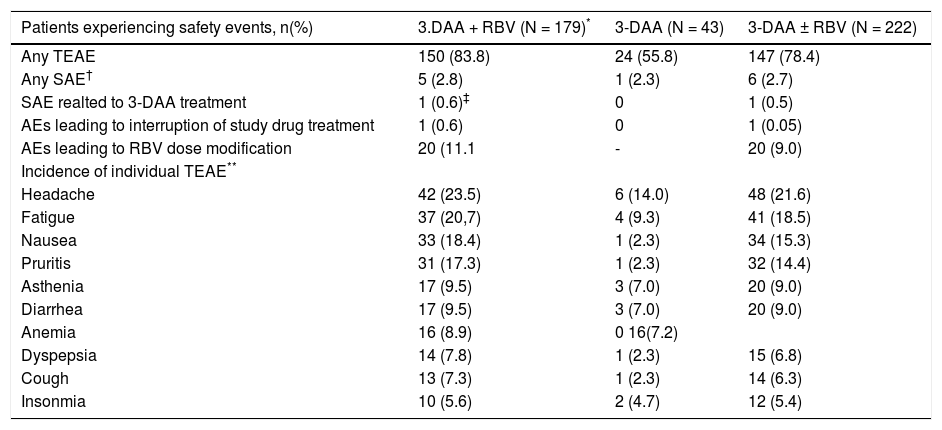
Among 222 patients in the safety population, 78.4% (174/ 222) patients experienced at least 1 TEAE, and 2.7% (6/222) patients experienced at least 1 serious AE (SAE) (Table 4). One (0.5%) cirrhotic patient assigned to 3-DAA plus RBV discontinued study drug treatment after Week 11 (Day 77) because of a TEAE (Grade 2 ascites, assessed by the investigator as having no reasonable possibility of being related to study drug), which started 5 days prior to discontinuation of study drug and resolved 28 days later. This patient achieved SVR12, but not SVR24 due to missing data, and subsequently developed HCC and died (see description below). TEAEs reported with an incidence ≥ 10% during treatment included headache (21.6%), fatigue (18.5%), nausea (15.3%) and pruritus (14.4%).
Adverse events.
| Patients experiencing safety events, n(%) | 3.DAA + RBV (N = 179)* | 3-DAA (N = 43) | 3-DAA ± RBV (N = 222) |
|---|---|---|---|
| Any TEAE | 150 (83.8) | 24 (55.8) | 147 (78.4) |
| Any SAE† | 5 (2.8) | 1 (2.3) | 6 (2.7) |
| SAE realted to 3-DAA treatment | 1 (0.6)‡ | 0 | 1 (0.5) |
| AEs leading to interruption of study drug treatment | 1 (0.6) | 0 | 1 (0.05) |
| AEs leading to RBV dose modification | 20 (11.1 | - | 20 (9.0) |
| Incidence of individual TEAE** | |||
| Headache | 42 (23.5) | 6 (14.0) | 48 (21.6) |
| Fatigue | 37 (20,7) | 4 (9.3) | 41 (18.5) |
| Nausea | 33 (18.4) | 1 (2.3) | 34 (15.3) |
| Pruritis | 31 (17.3) | 1 (2.3) | 32 (14.4) |
| Asthenia | 17 (9.5) | 3 (7.0) | 20 (9.0) |
| Diarrhea | 17 (9.5) | 3 (7.0) | 20 (9.0) |
| Anemia | 16 (8.9) | 0 16(7.2) | |
| Dyspepsia | 14 (7.8) | 1 (2.3) | 15 (6.8) |
| Cough | 13 (7.3) | 1 (2.3) | 14 (6.3) |
| Insonmia | 10 (5.6) | 2 (4.7) | 12 (5.4) |
AE: adverse event SAE: serious AE. TEAE: treatment-emergent AE.
Six patients (2.7%) experienced SAEs during the study (Table 4). A 65-year-old male patient with cirrhosis experienced hepatic decompensation starting at post-treatment Day 10 that was considered to be possibly related to treatment with both the 3-DAA regimen and RBV in the opinion of the investigator. This patient was classified as Child-Pugh B at screening, which was considered a protocol deviation. This patient also had a presumptive diagnosis of spontaneous bacterial peritonitis at this time (aerobic and anaerobic cultures of ascitic fluid were negative), and treatment with intravenous ceftriaxone was initiated. The event of hepatic failure was considered resolved 20 days after the last dose of study drug was administered. Additionally, a 49-year-old male patient without cirrhosis experienced renal failure and a transient ischemic attack on treatment Day 22. Both SAEs in the latter individual were considered to be possibly related to treatment with RBV, but not the 3-DAA regimen, in the opinion of the investigator. All other SAEs were not related to treatment in the opinion of the investigator.
The overall incidence of AEs and SAEs and the incidence of certain AEs were numerically higher in patients assigned to receive RBV than in patients assigned to the 3-DAA regimen alone, although AEs were generally of mild to moderate severity and clinically manageable (Table 4). In particular, anemia was not reported as an AE in any patient assigned to the RBV-free regimen, compared with 8.9% (16/ 179) of patients receiving the 3-DAA regimen with RBV.
Adverse events leading to RBV dose modification (as recommended in the RBV prescribing information) occurred in 20 (9.0%) of patients. These events included anemia (8 subjects), decreased hemoglobin (6 subjects), fatigue (4 subjects), asthenia, cough (2 subjects each), and decreased creatinine clearance, hypotension, pruritus, rash, renal failure, and respiratory tract infection (1 subject each).
Three (1.4%) deaths occurred during the post-treatment follow-up period > 4 months after taking the last dose of study drugs. One death due to HCC occurred 22 weeks post-treatment in a 54-year-old male patient with cirrhosis who had no evidence of HCC on baseline screening examinations; one death due to sepsis and hepatic decompensation occurred 21 weeks post treatment following complications related to total hip arthroplasty in a 59-year-old female patient with cirrhosis; and one death due to septicemia occurred 36 weeks post treatment in a 64-year-old male patient with diabetic vasculopathy, who developed an Acinetobacter infection after foot trauma. None of the three deaths were considered to be related to study drug treatment in the opinion of the investigator.
No patients experienced a Grade 3 or greater increase in ALT level during the study. One patient experienced a Grade 3 decrease in hemoglobin and one patient experienced a Grade 4 decrease in hemoglobin during 12 weeks of treatment with the 3-DAA + RBV regimen.
Twelve patients (5.4%) experienced Grade 3 elevations in total bilirubin concentration during treatment. All of these patients had cirrhosis and were assigned to the 3-DAA + RBV regimen. No patients experienced a Grade 4 increase in this parameter during the study.
DiscussionThe efficacy and safety of OBV/PTV/r + DSV (3-DAA) with and without RBV have been demonstrated in several previous large international phase 3 studies in patients with GT1 infection.10–15 The present study confirms the efficacy and safety profile of the 3-DAA regimen in Brazilian patients with advanced liver fibrosis including compensated cirrhosis, thrombocytopenia (platelets as low as 25,000/mm3), hypoalbuminemia (albumin as low as 2.8 g/ dL), and moderate renal dysfunction (CrCl as low as 30 ml/min). Overall, 96.4% of the patients in the ITT population achieved an SVR12. Efficacy rates were consistently high among GT1a-infected patients with (92.2%) and without (95.7%) cirrhosis, and among GT1b-infected patients with (100%) and without (97.7%) cirrhosis. Importantly, only 7 out of 222 patients (3.2%) experienced virologic failure when treated with the 3-DAA regimen. Furthermore, the 3-DAA regimen produced high response rates regardless of previous treatment history or advanced fibrosis staging. These results corroborate the findings of another clinical trial that explored the need for RBV among GT1b-infected patients with cirrhosis. In the TUR-QUOISE-III trial, 60 HCV GT1b-infected patients with cirrhosis were treated with the 3-DAA regimen without RBV.15 All 60 patients (100%) achieved an SVR12, indicating that RBV is not required to maximize
In resistance analyses, substitutions at resistance-associated amino acid positions in NS3, NS5A and or NS5B were assessed at baseline and at the time of virologic failure in the seven patients (6 GT1a, 1 GT1b) who experienced virologic failure. The small number of patients who failed to achieve SVR12, and the overall pattern of resistance observed in these individuals suggests that there was no correlation between the baseline resistance profile and the substitutions that emerged during treatment.
Treatment with the 3-DAA regimen was well tolerated in the present trial and the results of the safety analysis are consistent with that reported previously in patients with advanced hepatic fibrosis.14,15 In a population that included only patients with advanced liver fibrosis, no new safety signals were reported during the study. One patient discontinued treatment because of a TEAE (ascites that evolved into a diagnosis of HCC and death), and three patients died during follow-up (>4 months after the last dose of study drug treatment); however, none of these events was considered to be associated with study drug treatment. The incidence of AEs such as fatigue, nausea, pruritus and anemia were consistently higher in patients who received an RBV-containing regimen than in those receiving the 3-DAA regimen alone. These AEs are likely associated with RBV as they have previously been shown to be reported at a higher frequency among patients randomized to the 3-DAA regimen with RBV, compared to patients randomized to the 3-DAA regimen alone,11,13 and are also associated with RBV according to the drug package insert.16 Overall, the regimen was also well tolerated in patients with a wide range of comorbid conditions, such as hypertension, diabetes mellitus, depression, hypothyroidism, hypoalbuminemia, thrombocytopenia and moderate renal dysfunction. Anemia was not reported in patients assigned to the RBV-free regimen and the incidence of anemia was < 10% in patients who received RBV. Paritaprevir is a known inhibitor of the OAT1B1 bilirubin transporter, which is likely responsible for the isolated and largely asymptomatic increases in bilirubin serum levels that were observed in the present study.
A notable strength of the study is the inclusion of a large proportion of patients with advanced liver fibrosis (F3/F4) and comorbid conditions, such as thrombocytopenia, and moderate renal dysfunction. It must be emphasized however, that patients with a history of decompensated cirrhosis were not eligible for this study, and that the 3-DAA regimen should be avoided in patients with decompensated cirrhosis or a previous history of decompensation. Since patients with advanced fibrosis and a wide range of comorbidities are eligible for treatment under Brazil’s universal health program, the high overall efficacy and good tolerability demonstrated in this trial could be generalized to the population in which treatment is recommended in Brazil. The inclusion of only patients with advanced fibrosis also limits the generalizability of the results to a broader population that includes patients with stage F1 and F2 fibrosis; however, other studies have focused on treatment of patients with minimal liver fibrosis, or the full spectrum of fibrosis stages.10–13,17
This trial has certain limitations. The open-label design of the study is unlikely to have biased the reporting of efficacy results because HCV RNA is an objective laboratory assessment; however, when combined with the lack of control group it could have introduced bias in the reporting of safety events. However, the 3-DAA plus RBV regimen was well tolerated in two placebo-controlled trials in which the most common adverse events were fatigue, headache, and nausea, which is consistent with the results of the present trial.10,12
At the time the study was planned and initiated, few DAAs were approved and available for use in Brazil. IFN-based regimens could have been used as a comparator, however, it would have been difficult to blind patients to treatment with IFN-based regimens and patients may not have been willing to accept IFN-based therapy in a comparative trial because of the poor tolerability and low SVR12 rates. The study was also not powered for comparisons among subgroups of patients with different fibrosis staging or prior treatment history.
In conclusion, the results of this trial confirm that treatment with the 3-DAA regimen with RBV, or without RBV is safe and can be an effective option for the treatment of chronic HCV GT1 infection in the Brazilian patient population with bridging fibrosis or cirrhosis (METAVIR F3/4). In particular, the 3-DAA regimen without RBV is suitable for GT1b-infected patients without cirrhosis, whereas the 3-DAA regimen with RBV is required to maximize efficacy in GT1b patients with cirrhosis and all GT1a patients.
Abbreviations- •
AE: adverse event.
- •
ALT: alanine aminotransferase.
- •
APRI: AST to platelet ratio index.
- •
AST: aspartate aminotransferase.
- •
DAA: direct-acting antiviral agent.
- •
DSV: dasabuvir.
- •
EOT: end of treatment.
- •
GT1b: HCV genotype 1b.
- •
HCV: hepatitis C virus.
- •
LLOQ: lower limit of quantification.
- •
OBV: ombitasvir;
- •
PTV/r: paritaprevir coadministered with ritonavir.
- •
RBV: ribavirin.
- •
SAE: serious AE.
- •
SVR: sustained virologic response.
- •
TEAE: treatment-emergent adverse events.
- •
ULN: upper limit of normal.
The authors declares that there is no conflict of interest regarding the publication of this article.
Financial SupportAbbVie provided funding for this study and participated in design, research, data collection, interpretation of data, writing, reviewing and approving of the publication.
Financial Disclosure- •
MG Pessoa: Research grants: AbbVie, and Janssen; Advisory Board participation and speaker for: AbbVie, Alexion, Gilead, BMS, Janssen, and MSD.
- •
JVR Madruga: participation in clinical studies: AbbVie.
- •
K Alves, SA Lari, L Liu, R Tripathi, T Pilot-Matias, DE Cohen, NS Shulman: employees of AbbVie and may hold stock or stock options.
- •
EP Nunes: Speaker for, Advisory Board participation in, and Research grant from AbbVie.
- •
H Cheinquer: Research grants: AbbVie, BMS, Gilead, MSD; Advisory Board participation: AbbVie, BMS, Gilead, MSD; Speaker for: AbbVie, BMS, Gilead, MSD, Janssen.
- •
CE Brandāo-Mello: Speaker for and Consultant to AbbVie, Merck, Gilead, Janssen and BMS; Member: National and State of Rio de Janeiro Committee of Viral Hepatitis and Liver Diseases.
- •
CM Correa: Participation in clinical studies: AbbVie.
- •
ML Ferraz: Participation in clinical studies: AbbVie.
- •
P Ferreira: Advisory Board participation: AbbVie; Scientific Support: AbbVie.
- •
MR Álvares-da-Silva: Research grants: AbbVie, Gilead, MSD, Fiocruz, Eisai, and Janssen; Advisory Board participation: AbbVie, Bayer, Gilead, and MSD. Speaker for: AbbVie, Bayer, Gilead, MSD, and Janssen; Member: Ministry of Health Committees on Hepatitis C and Liver Transplant.
- •
HCS Moraes: Participation in clinical studies: AbbVie.
- •
E de Araujo: Participation in clinical studies: AbbVie.
- •
J Furtado: Participation in clinical studies: AbbVie.
- •
R Parana: Participation in clinical studies: AbbVie.
- •
G Silva: Advisory Board participation and Scientific Support: AbbVie.
- •
A Martinelli: Participation in clinical studies: AbbVie.
Medical writing assistance was provided by Blair Jarvis MSc, ELS of Medical Expressions, funded by AbbVie.



