



Endurance exercise (EXE) has emerged as a potent inducer of autophagy essential in maintaining cellular homeostasis in various tissues; however, the functional significance and molecular mechanisms of EXE-induced autophagy in the liver remain unclear. Thus, the aim of this study is to examine the signaling nexus of hepatic autophagy pathways occurring during acute EXE and a potential crosstalk between autophagy and apoptosis.
Materials and methodsC57BL/6 male mice were randomly assigned to sedentary control group (CON, n=9) and endurance exercise (EXE, n=9). Mice assigned to EXE were gradually acclimated to treadmill running and ran for 60min per day for five consecutive days.
ResultsOur data showed that EXE promoted hepatic autophagy via activation of canonical autophagy signaling pathways via mediating microtubule-associated protein B-light chain 3 II (LC3-II), autophagy protein 7 (ATG7), phosphorylated adenosine mono phosphate-activated protein kinase (p-AMPK), CATHEPSIN L, lysosome-associated membrane protein 2 (LAMP2), and a reduction in p62. Interestingly, this autophagy promotion concurred with enhanced anabolic activation via AKT-mammalian target of rapamycin (mTOR)-p70S6K signaling cascade and enhanced antioxidant capacity such as copper zinc superoxide dismutase (CuZnSOD), glutathione peroxidase (GPX), and peroxiredoxin 3 (PRX3), known to be as antagonists of autophagy. Moreover, exercise-induced autophagy was inversely related to apoptosis in the liver.
ConclusionsOur findings indicate that improved autophagy and antioxidant capacity, and potentiated anabolic signaling may be a potent non-pharmacological therapeutic strategy against diverse liver diseases.
The liver is one of the largest organs in our body and plays a key role in metabolism, immunity, and detoxification of harmful substances in the blood. Unlike other terminally differentiated organs such as heart and brain, the liver possesses a great regenerative and replicative capacity such that low degree of damage to the liver does not result in a major health problem. However, unresolved chronic stresses as a result of prolonged high-fat diet and medications along with extended sedentary life style has been known to undermine hepatic function, thus contributing to development of dysfunctional liver. Therefore, a healthy life style (e.g., good diet and regular physical activities) is important to maintain healthy liver. In this regard, regular endurance exercise (EXE) has been a non-pharmacological strategy to protect the liver against various liver diseases [1–3]. However, molecular mechanisms responsible for EXE-induced hepatic protection remain poorly understood. Recently, autophagy has emerged as a potential mechanism that help maintain cellular homeostasis in various tissues, as the absence of autophagy results in cellular injuries and even death including the heart, brain and liver [4–7]. Therefore, it is important to explore whether EXE promotes autophagy, and if so, elucidating signaling nexus of EXE-induced autophagy pathways is necessary to understand a potential mechanism of hepatic homeostasis in response to EXE.
Autophagy is a lysosome-dependent catabolic process by which potentially toxic molecules such as damaged proteins, lipids and dysfunctional small organelles are safely removed and recycled [8]. Any obstruction in the process of autophagy results in impairment of normal cellular function [9–12]; however, excessive autophagy (too much of good things) can lead to apoptotic cell death. Autophagy is induced under hypoxia [13,14] and nutrient deficiency [15,16], and growing evidence also shows that EXE is a strong inducer of autophagy in the heart and skeletal muscle [17,18]. Surprisingly, only a few studies reported that EXE improves hepatic autophagy; but even these observations were based upon the combinatory intervention of both EXE and high-fat diet without EXE only intervention [19,20]. Therefore, very little is known about hepatic signaling pathways induced by EXE per se. Moreover, because long-term EXE may alter general autophagy signaling due to exercise adaptation [21], establishment of acute exercise-induced autophagy signaling pathways is important to understand how endurance exercise induces hepatic autophagy.
Molecular signaling pathways of autophagy has been widely corroborated via several mechanistic studies [22,23]. For example, activation of adenosine monophosphate kinase (AMPK) phosphorylates Unc-51 like autophagy activating kinase1 at Ser555 (ULK1ser555), that subsequently activates a class III phosphatidylinositol 3 kinase (PI3K). This is an important initiation process for nucleation of phagophore formation [24]. Following the nucleation, a rate limiting enzyme of autophagy, ATG7 mediates translocation of activated microtubule-associated protein 1A/1B-light chain 3-II (LC3-II) to an elongated phagophore [25]. The LC3-II is then bound to targeted cellular cargos that are destined for degradation; thus, modulation of LC3-II levels has been used as a key marker of autophagy. Since autophagy is a lysosome-dependent degradation process, it has been suggested that a proportional increase in lysosomal biogenesis concurs with elevated LC3-II levels [26]. In line with these reports, upregulation of lysosomal proteins such as lysosome-associated membrane protein 2 (LAMP2) and a protease CATHEPSIN have been observed in numerous studies. Also, studies have demonstrated that a transcription factor EB (TFEB) plays a crucial role in LAMP2 and CATHEPSIN [27,28].
While autophagy promotion has been linked to cellular survival and health, excessively upregulated autophagy can also mediate cell death. For example, when autophagy is overly activated, cell death (e.g., apoptosis) concur, evidenced by an increase in TUNEL positive cells, cysteine-dependent aspartate-directed protease 3 (CASPASE3) activation, cleavage of poly ADP ribose polymerase (PARP), and upregulation of oncogenes (e.g., p53 and p21). Currently, it is unknown whether EXE-induced autophagy upregulation coincides with or suppresses apoptosis. In this study, we investigated molecular signaling nexus of short-term EXE-induced autophagy in the liver and examined potential relationship between autophagy and apoptosis in response to EXE.
2Material and methods2.1AnimalsMales C57BL/6 mice (age: 9 weeks) were purchased from ENVIGO (Indianapolis, IN), housed in an animal facility at 12h light:12h dark cycle, and fed with a standard chow diet ad libitum with free access to water. We complied with the rule of the Guide for the Care and Use of Laboratory Animals (1996, published by National Academy Press, 2101 Constitution Ave. NW, Washington, DC 20055, USA), and all procedures required in this study were approved by the Institutional Animal Care and Use Committee (approval number: 2017-004). After one-week of environment acclimation, the animals were randomly assigned to two groups: a sedentary control (CON, n=9) group and an endurance exercise (EXE, n=9) group.
2.2Treadmill running exercisePrior to treadmill exercise, mice assigned to a EXE group were familiarized with running on a motorized animal treadmill for 30min/day for five days, with daily running speed gradually increased up to 12m/min at the last day of acclimation, while mice assigned to a CON group remained in their cage. After five days of familiarization with treadmill running, the EXE group performed five days of treadmill running exercise, starting with 10min warming up at a speed of 10m/min on a 0% grade after which the speed was increased at 15m/min and maintained for 60min. The efficacy of this treadmill exercise has been demonstrated in our previous study [17]. To preclude possible confounding results that may be caused by electrical shocks, we did not use electrical grids but instead applied soft plastic brushes at the end of each lane. Animals touching the brush become aroused and continued running. Animals failing to run despite continuous touching with the brush were allowed to terminate their exercise to eliminate undesired stress responses.
2.3Tissue collection and storage90min after the last exercise session, animals were sacrificed by cervical dislocation, and liver tissues were immediately excised and washed with ice-cold PBS solution to remove remaining blood. Then, the tissue samples were collected, covered with optimal cutting temperature (OCT) freezing medium, and frozen in isopentane pre-cooled with liquid nitrogen. The rest of tissues were wrapped in aluminum foil and immediately frozen in liquid nitrogen and stored in −80°C until needed.
2.4Immunofluorescence microscopyThe tissue preparation for immunohistochemistry was based on our published work [29]. Briefly, frozen liver tissues were cryo-sectioned (10μm) with a sliding cryotome (Leica, Germany) were fixed with 4% paraformaldehyde on ice for 15min, rinsed with PBS (pH 7.4), and then blocked with 10% normal goat serum for 1h. A LC3A/B antibody (1:200) was applied on the tissue sections and incubated overnight at 4°C. Next day, the sections were washed with PBS and incubated for 1h at room temperature with secondary antibodies (Alexa 488-conjugated goat anti-rabbit, ThermoFisher, USA). After the tissue sections were washed with PBS, nuclei were stained with Hoechst 33342. Then, the sections were mounted on cover slides with Vectashield (Vector Laboratories, CA), and digital images were captured at 40X magnification using a fluorescence microscope (EVOS, ThermoFisher, USA). The number of LC3-positive puncta on the images was manually counted (15 images per tissue).
2.5Western blottingTotal protein extraction from the liver tissues was based on our published studies [21,29]. Briefly, the liver tissues were homogenized with glass homogenizer in T-PER® tissue protein extraction reagent (ThermoFisher Scientific, USA) containing a Halt™ Protease and Phosphatase inhibitor cocktail (ThermoFisher, Scientific USA), incubated on ice for 30min, and centrifuged at 20,817×g (5804R, FA-45-30-11, Eppendorf, Germany) for 20min to extract proteins. The extracted proteins from the tissues were equally normalized based up on the Bradford protein assay and prepared for SDS-PAGE. Proteins were separated by 10% NuPAGE™ Bis-Tris Gel (Life Technology, USA) and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat milk for non-phospho proteins or 5% bovine serum albumin in Tris-buffered saline solution containing 0.1% Tween 20 (TBST) for 1h at room temperature, after which the membranes were incubated over night at 4°C with designated primary antibodies. The primary antibodies were as follows: AKT (#9272, 1:1000), AMPKα (#2532, 1:1000), ATG 7 (#2631, 1:1000), BECLIN-1 (#3738, 1:1000), BNIP3 (#3769, 1:1000), LC3A/B (#12741, 1:1000), MnSOD (#13194, 1:1000), mTOR (#2972, 1:1000), PARP (#9532, 1:1000), phospho-AKT at Ser473 (#9271, 1:1000), phospho-AMPKα at thr172 (#2535, 1:1000), phospho-mTOR at Ser2481 (#2974, 1:1000), SQSTM1/p62 (#5114, 1:1000), phospho-ULK-1 at Ser757 (#14202, 1:1000), and ULK-1 (#8054, 1:1000) from Cell Signaling (Danvers, MA); CATHEPSIN L (ab58991, 1:1000) and TFEB (ab2636, 1:1000) from Abcam (Cambridge, MA); BCL-2 (sc-492, 1:1000), phospho-BCL-2 (sc-377576, 1:1000), GPX1/2 (sc133160, 1:1000), PGC-1α (sc-13067, 1:1000), p70S6Kα (sc-8418, 1:1000), phospho-p70S6Kα (sc-8416, 1:1000), p21 (sc-6246, 1:1000), p53 (sc-393031, 1:1000), and PRX3 (sc-23973, 1:1000) from Santa Cruz Biotechnology (Santa Cruz, CA); LAMP2 (PA1-655, 1:1000) from ThermoFisher Scientific (Rockford, IL); CASPASE3 (NB100-56112SS, 1:1000) and CuZnSOD (NBP2-24915, 1:5000) from NOVUS biological (Littleton, CO); phospho-ULK-1 at Ser555 (#ABC124, 1:1000) from Millipore (Temecula, CA). After washing off primary antibodies with TBST, the membranes were incubated with designated secondary antibodies (goat anti-mouse or anti-rabbit HRP conjugated: Life Technology, USA) for 1h at room temperature and washed with TBST. Digital blot images of target proteins were acquired using the ECL Western blotting detection substrates (GE Healthcare, USA) and a ChemiDoc XRS imaging system (Bio-Rad, USA). The intensity of target protein was analyzed and quantified with an Image Lab Software (Bio-Rad, CA). Each target protein intensity was normalized by the intensity of Ponceau-stained proteins, and all protein levels were presented as fold changes.
2.6Statistical analysisAll values were expressed as means±standard error of the mean (SEM). Data shown in bar graphs were based upon fold changes compared to CON group. For statistical analysis, a student t-test (unpaired, one tail) was executed using a Prism 6 software (GraphPad, USA) to identify statistical significance between groups. Statistical significance was set at p<0.05.
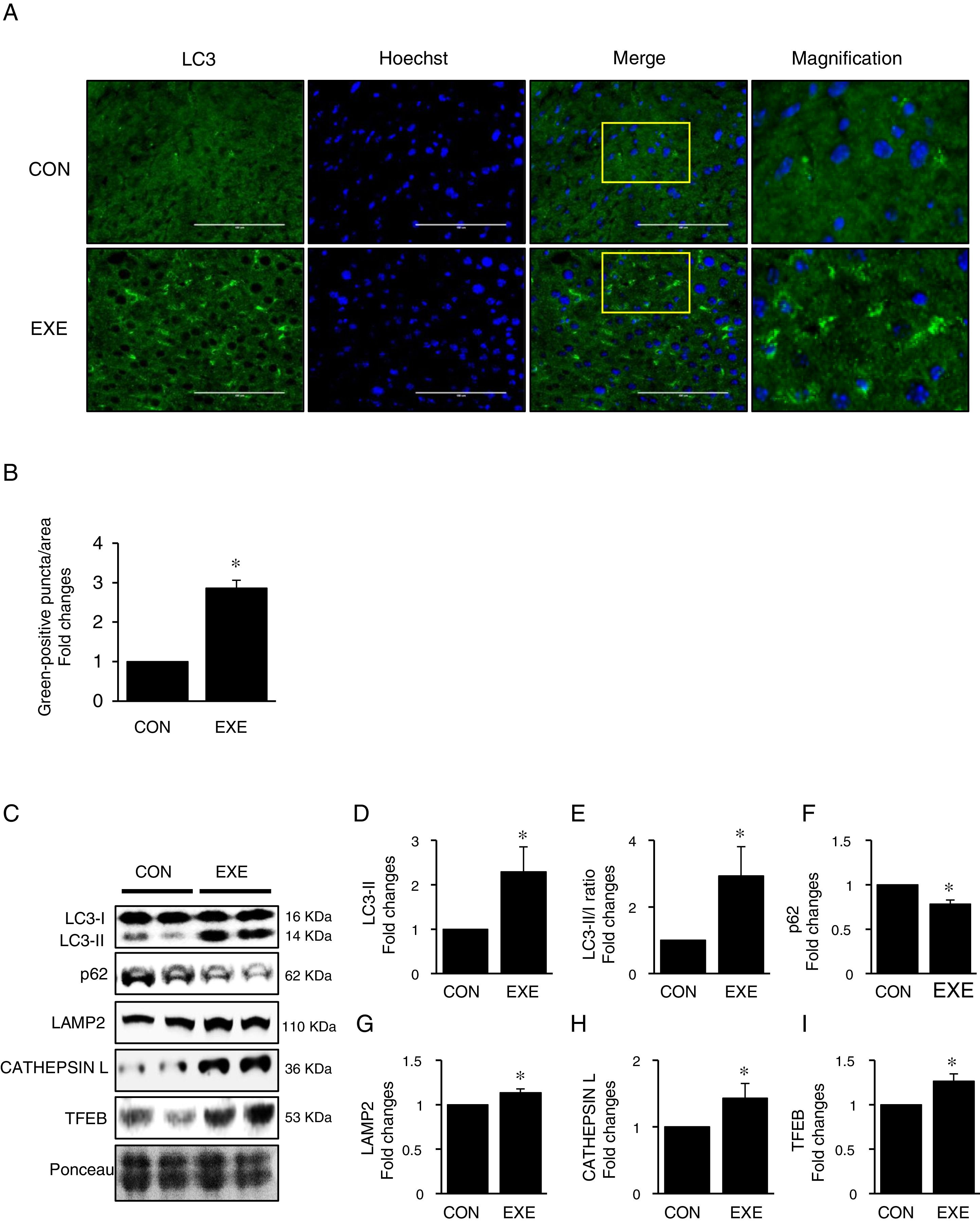
3Results3.1Endurance exercise promotes autophagy flux and a lysosomal biosynthesisAutophagy promotion is typically confirmed by an increase in LC3-II levels or a LC3-II/I ratio. Thus, to examine if EXE enhances hepatic autophagy, we assessed both LC3-II contents and the ratio of LC3-II to LC3-I in liver tissues of EXE-trained and sedentary animals. We found that EXE-trained animals significantly elevated LC3 positive puncta in the cryo-sectioned liver tissues examined by immunofluorescence microscopy (Fig. 1A and B). In addition, the EXE group displayed elevated LC3-II levels as well as LC3-II-/I ratio, compared to CON group (Fig. 1C–E). Since disruption of a fusion process between autophagosomes and lysosomes rather than truly enhanced autophagy flux can lead to LC3-II accumulation and thus misinterpretation of autophagy flux, we measured an autophagy adaptor protein p62 that is irreversibly degraded by lysosomes and has been recommended for the measurement of autophagy flux [30]. With reduction in p62 with LC3-II elevation suggesting enhanced autophagy flux, we observed that EXE resulted in the decline of p62 levels (Fig. 1C and F). In addition, since an increase in lysosomal elements contributes to facilitating autophagy flux, we measured lysosomal proteins (LAMP2 and CAPTHEPSIN L) and a key lysosomal transcription factor, TFEB and found that these proteins were upregulated in response to EXE (Fig. 1C and G–I).
 Representative images of fluorescence microscopy showing accumulation of autophagosomes. Green color presents LC3-positive puncta. (B) Quantitative assessment of autophagosomes (numbers of green fluorescent puncta/area) (n=3 per group). (C) Representative images displaying protein expressions. Liver tissue homogenates were immunoblotted for LC3-I, LC3-II, p62, LAMP2, CATHEPSIN L, and TFEB. (D–I) Quantification of proteins listed in (C) (n=9 per group). Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Each target protein was normalized by the loading control. Data are presented as a mean±SEM. * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.")
Endurance exercise enhances hepatic autophagy and lysosomal biogenesis. (A) Representative images of fluorescence microscopy showing accumulation of autophagosomes. Green color presents LC3-positive puncta. (B) Quantitative assessment of autophagosomes (numbers of green fluorescent puncta/area) (n=3 per group). (C) Representative images displaying protein expressions. Liver tissue homogenates were immunoblotted for LC3-I, LC3-II, p62, LAMP2, CATHEPSIN L, and TFEB. (D–I) Quantification of proteins listed in (C) (n=9 per group). Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Each target protein was normalized by the loading control. Data are presented as a mean±SEM. * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.
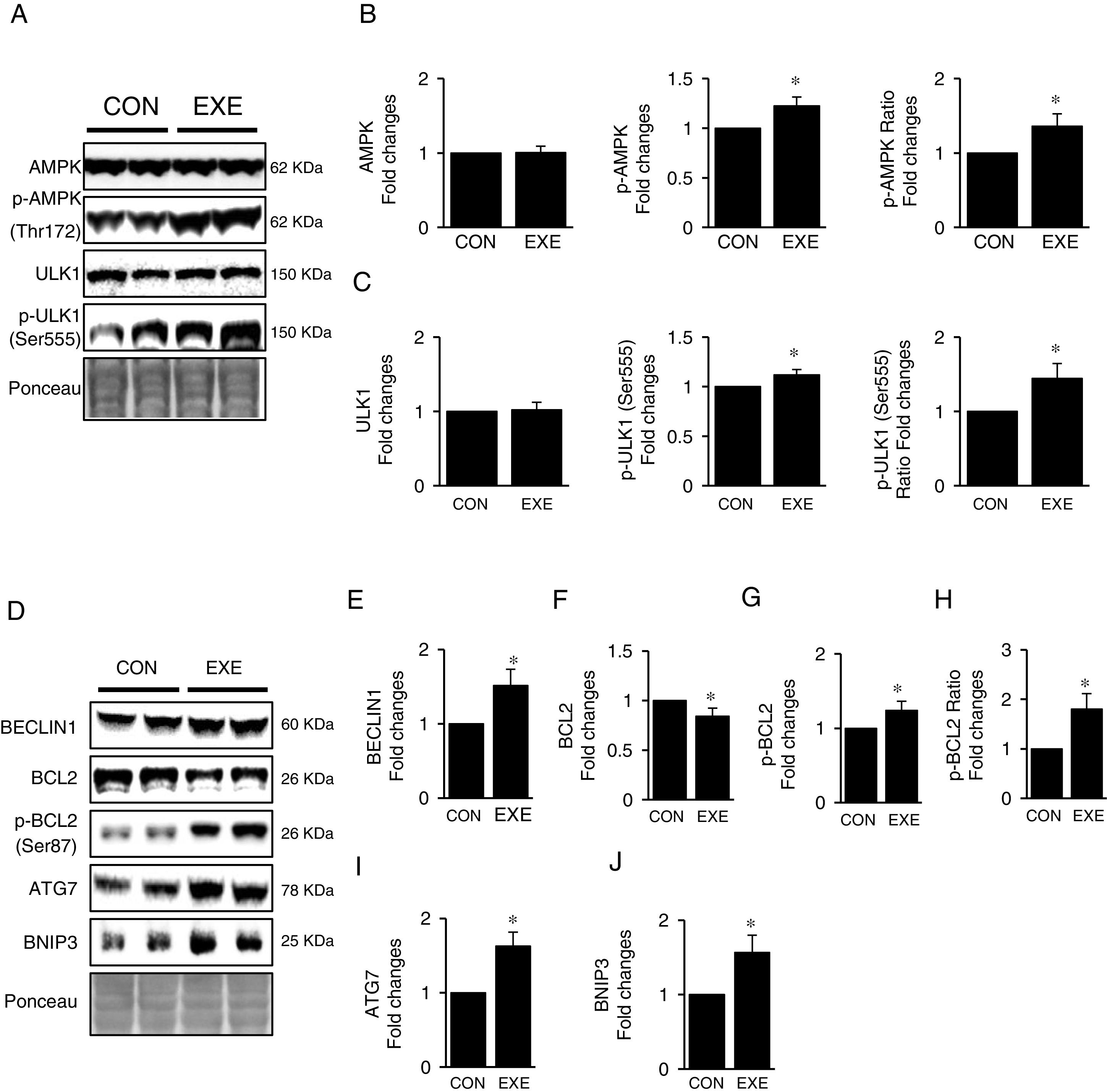
Next, we attempted to examine molecular signaling pathways of EXE-induced hepatic autophagy. Our data showed that EXE elevated phosphorylation levels of AMPK without modulating total AMPK levels (Fig. 2A and B). We next measured the phosphorylation state of ULK1, which is a downstream target of AMPK and found that ULK1 phosphorylation was slightly higher (approximately 15–20%) in response to EXE, compared to those in CON (Fig. 2A and C).
 Representative images of protein expressions. Liver tissue homogenates were immunoblotted for AMPK, p-AMPKThr172, and ULK1, p-ULK1Ser555. (B) Quantification of phosphorylation levels of p-AMPK and ratio of p-AMPK/total AMPK. (C) Quantification of phosphorylation levels of p-ULK1 and ratio of p-ULK1/total ULK1. (D) Representative images of protein expressions via immunoblotting for BECLIN1, BCL2, p-BCL2Ser87, ATG7, and BNIP3. (E) Quantification of BECLIN1. (F-H) Quantification of phosphorylation levels of p-BCL2 and ratio of p-BCL2/total BCL2. (I and J) Quantification of ATG7 and BNIP3, respectively. Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Each target protein was normalized by the loading control. Data are presented as a mean±SEM (n=9). * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.")
Inductive process of endurance exercise-induced autophagy resembles canonical signaling pathways. (A) Representative images of protein expressions. Liver tissue homogenates were immunoblotted for AMPK, p-AMPKThr172, and ULK1, p-ULK1Ser555. (B) Quantification of phosphorylation levels of p-AMPK and ratio of p-AMPK/total AMPK. (C) Quantification of phosphorylation levels of p-ULK1 and ratio of p-ULK1/total ULK1. (D) Representative images of protein expressions via immunoblotting for BECLIN1, BCL2, p-BCL2Ser87, ATG7, and BNIP3. (E) Quantification of BECLIN1. (F-H) Quantification of phosphorylation levels of p-BCL2 and ratio of p-BCL2/total BCL2. (I and J) Quantification of ATG7 and BNIP3, respectively. Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Each target protein was normalized by the loading control. Data are presented as a mean±SEM (n=9). * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.
BECLIN-1 is an important protein involved in an initial step in phagophore formation, and its dissociation from BCL2-BECLIN1 complexes upon BCL2 phosphorylation is also critical for autophagy. Our data confirmed that EXE not only upregulated BECLIN1 proteins (Fig. 2D and E) but also remarkably elevated BCL2 phosphorylation levels as well as p-BLC2/t-BLC2 ratio, despite reduced levels of total BCL2 proteins (Fig. 2D and F–H). Since ATG7 and BNIP3 have been suggested to enhance autophagy, we analyzed these proteins and observed that both ATG7 and BNIP3 were upregulated in response to EXE (Fig. 2D, I and J).
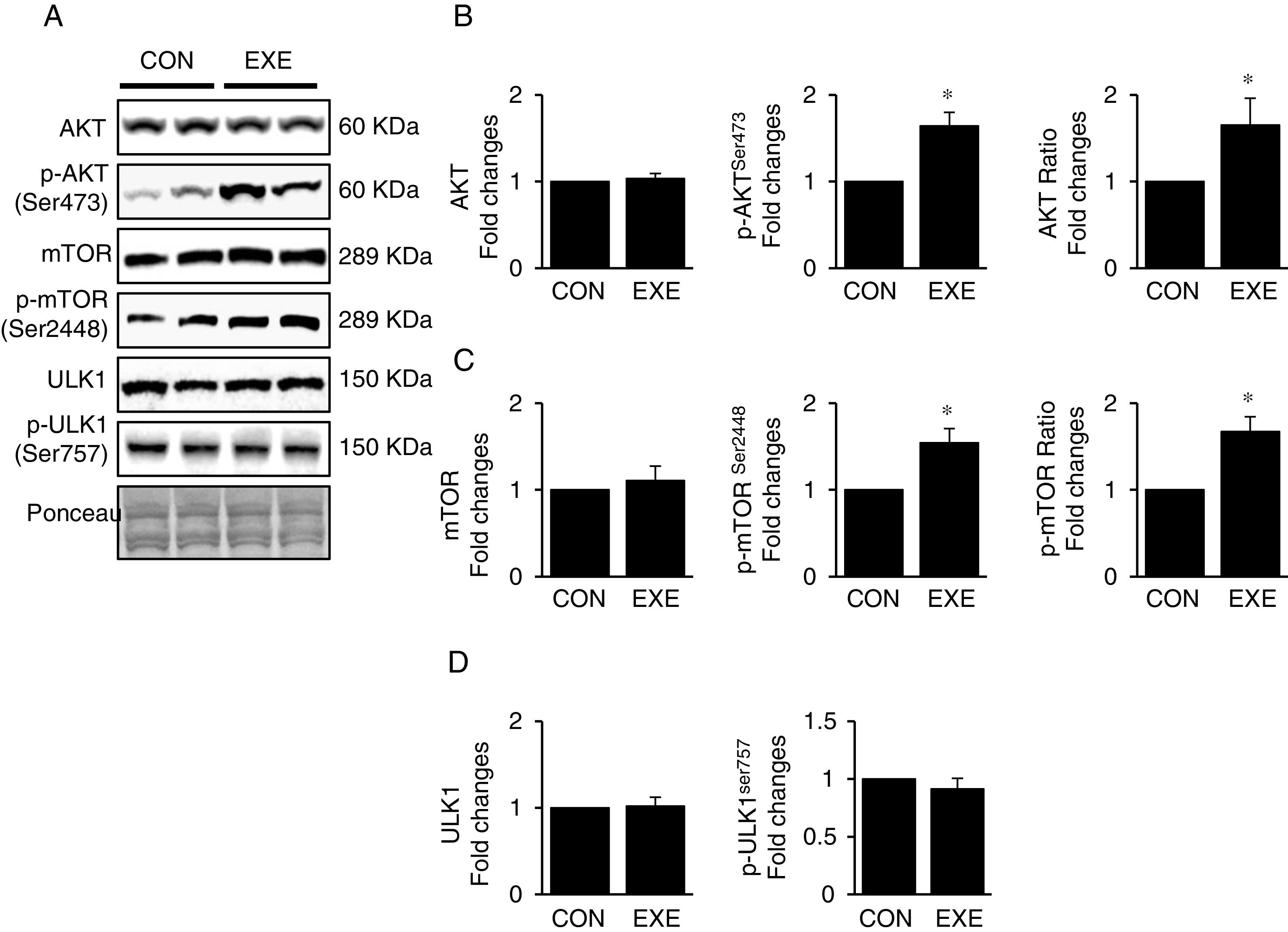
3.3Exercise-induced autophagy concurs with anabolic activationEndurance exercise has been reported to stimulate potentiation of anabolic signaling cascades (e.g., AKT-mTOR axis) in several different tissues [31,32]. In general, activation of this signaling interferes in autophagy, but some studies show EXE-induced autophagy in parallel with anabolic activation in hearts. Currently, whether this phenomenon is universally observed in liver remains unknown. Our data revealed that EXE significantly raised phosphorylation levels of AKT and p-AKT/t-AKT ratio without alteration of t-AKT (Fig. 3A and B) as wells as its downstream target, mTOR (Fig. 3A and C). We further examined if activated mTOR induces phosphorylation of its downstream target, ULK1 at Ser757 that is known to hinder autophagy induction. Intriguingly, with mTOR activation present, ULK1 phosphorylation levels were unchanged in response to EXE (Fig. 3A and D).
 Representative images of protein expressions. Liver tissue homogenates were immunoblotted for AKT, p-AKTSer473, and mTOR, p-mTORSer2448, ULK1, and p-ULK1Ser757. (B) Quantification of total AKT, phosphorylation levels of p-AKT and ratio of p-AKT/total AKT. (C) Quantification of total mTOR, phosphorylation levels of p-mTOR, and ratio of p-mTOR/total mTOR. (D) Quantification of ULK1and phosphorylation levels of p-ULK1Ser757. Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Each target protein was normalized by the loading control. Data are presented as a mean±SEM (n=9). * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.")
Endurance exercise potentiates the anabolic state. (A) Representative images of protein expressions. Liver tissue homogenates were immunoblotted for AKT, p-AKTSer473, and mTOR, p-mTORSer2448, ULK1, and p-ULK1Ser757. (B) Quantification of total AKT, phosphorylation levels of p-AKT and ratio of p-AKT/total AKT. (C) Quantification of total mTOR, phosphorylation levels of p-mTOR, and ratio of p-mTOR/total mTOR. (D) Quantification of ULK1and phosphorylation levels of p-ULK1Ser757. Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Each target protein was normalized by the loading control. Data are presented as a mean±SEM (n=9). * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.
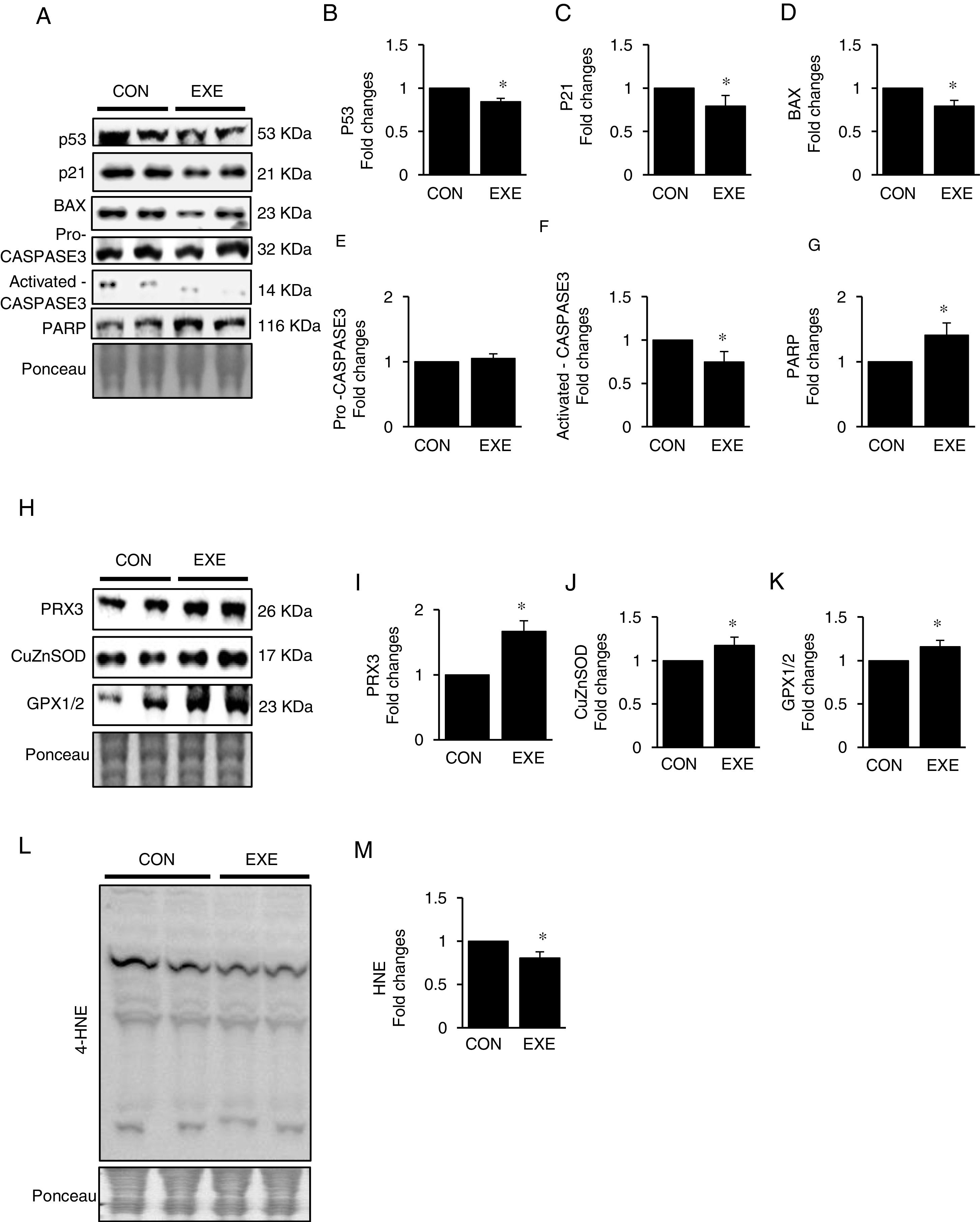
Autophagy is an essential avenue to sustain favorable cellular hemostasis; however, too much of autophagy is also linked to cell death. In order to determine whether EXE-induced autophagy promotion is beneficial or detrimental to liver tissues, we examined pro-apoptotic signaling pathways. EXE resulted in downregulation of pro-apoptotic proteins p53, p21, and BAX (Fig. 4A–D). Also, a cell death executioner, CASPASE3 (cleaved or active form) levels were diminished, whereas levels of an intact form of DNA repairing enzyme, PARP remained upregulated (Fig. 4A, and E–G). We next measured levels of endogenous antioxidant enzymes because improved antioxidant capacity is critically linked to protection of cell from apoptosis. EXE elevated levels of mitochondrial antioxidant enzyme, PRX3, and cytosolic antioxidant enzymes, CuZnSOD, and GPX, respectively (Fig. 4H–K). Supporting the increased antioxidant capacity, EXE reduced lipid peroxidation in the liver of EXE-trained animals, compared to that of CON animals (Fig. 4L and M).
 Representative images of protein expressions. Liver tissue homogenates were immunoblotted for apoptotic proteins p53, p21, BAX, pro-CASPASE3, activated CASPASE3, and DNA repairing enzyme PARP. (B–G) Quantification of proteins listed in (A). (H) Representative images of antioxidative proteins (PRX3, CuZnSOD, and GPX1/2) acquired by immunoblotting. (I–K) Quantification of PRX3, CuZnSOD, and GPX1/3, respectively. (L) A representative image of lipid peroxidation. Liver tissue homogenates were immunoblotted for 4-hydroxynonenal (HNE), an oxidative stress marker of lipid membrane. (M) Quantification of HNE. Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Data are presented as a mean±SEM (n=9). * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.")
Endurance exercise suppresses apoptosis and improves antioxidative capacity. (A) Representative images of protein expressions. Liver tissue homogenates were immunoblotted for apoptotic proteins p53, p21, BAX, pro-CASPASE3, activated CASPASE3, and DNA repairing enzyme PARP. (B–G) Quantification of proteins listed in (A). (H) Representative images of antioxidative proteins (PRX3, CuZnSOD, and GPX1/2) acquired by immunoblotting. (I–K) Quantification of PRX3, CuZnSOD, and GPX1/3, respectively. (L) A representative image of lipid peroxidation. Liver tissue homogenates were immunoblotted for 4-hydroxynonenal (HNE), an oxidative stress marker of lipid membrane. (M) Quantification of HNE. Ponceau-stained proteins on the nitrocellulose membrane were used as an internal control to ensure equal loading. Data are presented as a mean±SEM (n=9). * Indicates a statistical difference, compared to CON (p<0.05). Con: sedentary control, EXE: Endurance Exercise, SEM: standard of the mean.
EXE has been recognized as a potent inducer of autophagy in various tissues including skeletal muscle [33], heart [34], and brain [35]; surprisingly, very limited data are available about EXE-induced liver autophagy. In the present study, we demonstrate a remarkable increase in hepatic autophagy in response to short-term EXE and establish its detailed signaling pathways. In addition, our data show possible functional significance of EXE-induced autophagy in the liver since increased autophagy concurs with suppressed apoptosis in conjunction with enhanced antioxidant capacity.
Previous studies have reported that an acute bout of 60min of moderate intensity EXE serves as a strong inducer of autophagy in the heart and skeletal muscle [17,36,37]; however, it was very interesting that we did not observe any changes in autophagy levels in the liver with the acute bout of EXE protocol used in above studies; more interestingly, autophagy levels were not modulated up to four consecutive days of EXE (data not shown). We started observing upregulation of autophagy at the fifth day of EXE. Therefore, in this study, we chose five consecutive days of EXE to examine acute-EXE hepatic autophagy signaling pathways.
An increase in LC3-II is considered as a key indicator of enhanced autophagy in many studies including our present study. However, since LC3-II can be accumulated upon dysfunctional autophagy (e.g., defects in lysosomal fusion with autophagosomes or in proteolytic process) rather than through truly improved autophagy flux, additional measurement such as p62 has been recommended. For example, since p62 binds to cellular cargo molecules as well as to LC3-II and is degraded by lysosomes [38,39], diminished p62 levels in parallel with elevated LC3-II levels have been considered as an indicative of bona fide autophagy flux in most studies [40,41]. Consistent with these studies, our data also showed a reduced p62 level in the presence of LC3-II upregulation in the liver of EXE-trained animals, suggesting that EXE-mediated autophagy may not be due to interrupted autophagy process but to enhanced autophagic flux. Aside from p62, upregulation of lysosomal proteins such as LAMP2 [42,43] and CATHEPSIN L [44] strongly correlate with enhanced autophagy flux. Supporting this notion, our data showed upregulation of LAMP2 as well as CATHEPSIN L levels in EXE-trained animals concurrent with elevated LC3-II levels. To further explore the mechanism responsible for EXE-mediated upregulation of the lysosomal proteins, we next examined the effect of EXE on TFEB because this transcription factor has been identified as a master transcription regulator of most lysosomal proteins [45,46]. Our findings show that EXE upregulates TFEB levels, and this increase is associated with lysosomal protein overexpression. Importantly, given a recent exquisite study demonstrating that activated (dephosphorylated) TFEB by a phosphatase CALCINEURIN translocates to nucleus and initiates transcription of lysosome-related genes [45], it is important to elucidate in future studies if post-translational modifications of TFEB is essential for the EXE-mediated lysosomal. Taken together, our findings suggest that enhanced hepatic autophagy in response to EXE may be due to the accelerated induction of autophagy as well as the elevated lysosomal degradation. However, since the present did not examine whether the rate of autophagy flux (more accumulation of autophagosomes) increases in the presence of lysosome inhibitor chloroquine in response to EXE, future studies using a chloroquine are warranted to prove a definite phenomenon of EXE-induced autophagy.
While EXE has been recognized as a potent inducer of autophagy in various tissues, signaling nexus of EXE-induced autophagy pathways in the liver remains poorly understood. Multiple studies have revealed that activation (phosphorylation) of AMPK and its downstream kinase ULK1Ser555 is an essential step in autophagy induction [47,48]. Our data provide important evidence that EXE-mediated autophagy is also linked to AMPK-ULK1 activation, suggesting that AMPK phosphorylation is a crucial factor for induction of EXE-mediated autophagy. Our findings are also consistent with other studies showing a critical association of AMPK phosphorylation with autophagy in response to EXE in different tissues such as the heart and brain [17,36] In contrast to our study, a study by Alex et al. shows conflicting results that three weeks of EXE does not alter AMPK activities, resulting in no autophagy in the liver [49]. This discrepant observation may be explained by two main differential factors: (1) our study uses five-day consecutive days of EXE rather than three weeks; and (2) we collect tissue samples 1.5h after the last session of EXE rather than 24h. Of the two, the tissue collection time seems to be a primary factor that generates the opposing observation, given recent studies demonstrating a gradual decline in autophagy as the time of tissue collection elapses after reaching maximum levels between 60min and 90min post exercise in other tissues [17,36]. Further studies are necessary to verify the phenomenon in the liver.
In addition to AMPK potentiation, a recent study by He et al. reveals the essential role of BECLIN1 in EXE-induced autophagy [36]. For example, the authors show that inhibition of dissociation of BECLIN1 from a BCL2-BECLIN1complex by hindering BCL2 phosphorylation completely abolished EXE-induced autophagy, suggesting BCL2 phosphorylation is necessary for BECLIN1 to participate in EXE-induce autophagy in heart and skeletal muscles. Currently, no studies have been conducted yet as to whether the observed results occur in the liver in response to EXE. Our study, for the first time, shows that EXE promotes BCL2 phosphorylation and BECLIN1 upregulation, supporting the notion above that BCL2 phosphorylation would be substantial in the liver as well for the EXE-induced autophagy. Moreover, BNIP3 and ATG7 have been identified as key inducers of EXE-induced autophagy in various tissues [18,50]. Our present study supports the current literature by providing evidence of elevated levels of BNIP3 and ATG7 in the liver in response to EXE. These results suggest that potentiation of inductive processes of autophagy as revealed in other studies is important for EXE-mediated autophagy in the liver.
In general, enhanced anabolic signaling via activation of AKT-mTOR anabolic axis interrupts autophagy induction; for instance, activation of mTOR by AKT retards autophagy induction via mTOR's ability to phosphorylate ULK at Serine757, whereas inhibition of mTOR via rapamycin promotes autophagy [51–53]. Surprisingly, despite an increase in EXE-induced autophagy, phosphorylation levels of mTOR concurs, suggesting that EXE-induced autophagy occurs independent of anabolic activation. Supporting our observation, a recent study led by Lee at al. shows similar results in the heart of EXE-trained animals [17]. Currently, no studies are available to explain how EXE bypasses anabolic activation-induced autophagy suppression and upregulates autophagy. Evidently, finding a new regulatory switch of autophagy by revealing mechanisms of EXE-induced autophagy would be an interesting topic in future studies.
Our present study reveals that EXE significantly increases autophagy in the liver and provides detailed pathways of EXE-induced autophagy. However, in light of recent studies showing the chronic upregulation of autophagy is rather harmful and even a critical source of apoptotic cell death [54,55], it seems critical to examine whether enhanced autophagy by EXE is beneficial or detrimental to hepatocytes. In this regard, our study, for the first time, provides important evidence that EXE-induced autophagy confers beneficial effects in that EXE maintains lower levels of apoptotic signaling molecules; for example, EXE downregulates pro-apoptotic proteins such as p53, p21, and BAX. It is completely unknown how EXE represses these protein expression, but according to several studies showing p53 is a target of a E3 ubiquitin ligase, Mdm2 [56,57], we assume that EXE may increase a proteolytic system (e.g., ubiquitin-proteasome) via Mdm2 for the degradation of p53 or suppress its transcription. In addition to the suppressed pro-apoptotic protein levels, EXE also exhibits significantly lower levels of active (cleaved) forms of CASPASE3.
To further understand how EXE maintains low levels of apoptosis, we sought to examine antioxidant capacity since oxidative stress has been known to be linked to apoptosis. Growing evidence demonstrates that mitochondria [58,59] and NADPH oxidase [60,61] are major sources of superoxide anion (a free radical) production in the liver. However, endogenous antioxidant system prevents unfavorable oxidative stress; for example, superoxide anion molecules produced from mitochondria and NADPH oxidase are initially converted to a mild oxidant, hydrogen peroxide (H2O2) molecule by a manganese superoxide dismutase (MnSOD) [62,63] or a copper zinc dismutase (CuZnSOD) [64]. Then, H2O2 is detoxified by other antioxidant enzymes such as peroxiredoxin 3 (PRX3) [65,66] in mitochondria and glutathione peroxidases (GPX) in cytoplasm [67]. Our study shows that EXE upregulates endogenous antioxidant levels associated with both mitochondria (e.g., PRX3) and cytosol (e.g., CuZnSOD and GPX1/2) in parallel with reduced levels of lipid peroxidation. Consistent with our findings, other studies have also reported that EXE improves hepatic antioxidant capacity [68,69]. By contrast, other studies have shown no changes or even reductions in antioxidant levels after EXE [70,71]. Unfortunately, clear delineation of these discrepant results is not available yet, but several possible factors such as duration (short-term vs. long-term), modes (treadmill running exercise vs. swimming), and time of sacrifice (1h vs. 24–48h post exercise) may affect the status of antioxidant levels.
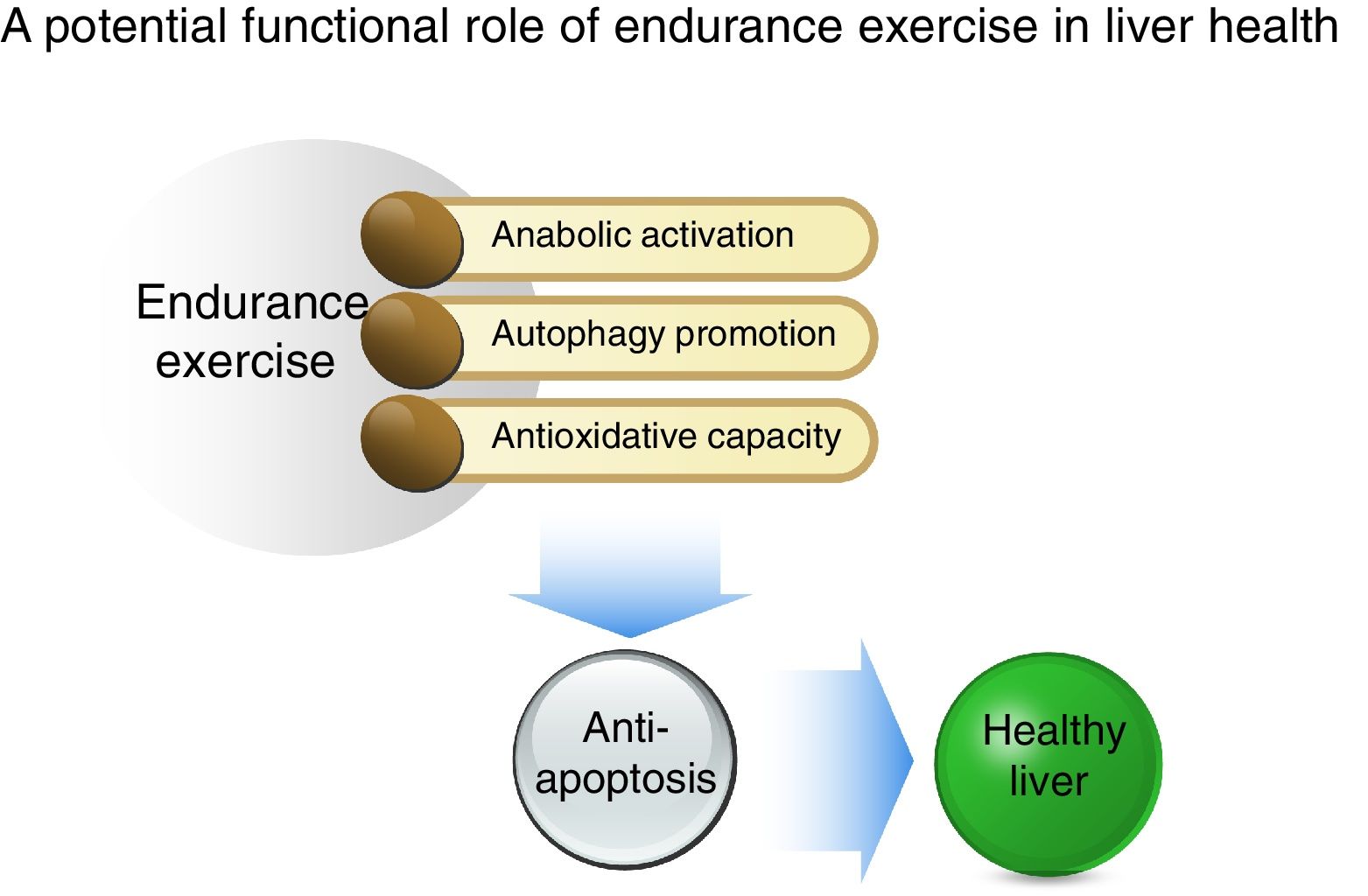
In conclusion, as illustrated in Fig. 5, our study shows that acute EXE results in enhanced hepatic autophagy during the early recovery periods and that EXE-induced autophagy coincides with activation of anabolic signaling (AKT-mTOR) and suppression of anti-apoptosis. Moreover, we report that EXE promotes antioxidative capacity. Taken together, our study suggests that EXE-induced autophagy and improved antioxidative capacity prohibits unnecessary apoptosis and thus provides suitable cellular environment. This favorable cellular adaptation acquired by regular EXE may be a critical underlying mechanism necessary for maintenance of a healthy liver.AbbreviationsEXE endurance exercise Unc-51 like autophagy activating kinase phosphatidylinositol 3 kinase lysosome-associated membrane protein 2 autophagy protein 7 microtubule-associated protein B-light chain 3 II adenosine mono phosphate-activated protein kinase mammalian target of rapamycin copper zinc superoxide dismutase glutathione peroxidase peroxiredoxin 3 transcription factor EB B-cell leukemia/lymphoma 2 BCL2 associated X protein cysteine-dependent aspartate-directed protease 3 poly ADP ribose polymerase

Summary of a potential protective mechanism of endurance exercise in the liver. Potentiated anabolic signaling, enhanced autophagy, and improved antioxidant capacity as a result of endurance exercise ensue in anti-apoptosis, suggesting that the reshuffle of favorable cellular environment by endurance exercise may be a critical source for hepatic protection.
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
FundingThis project was supported by a research grant from the University of West Florida though Office of Research and Sponsored Programs (R0062) and UWF Florida Research Fellowship to YL (CF6672 and CR0070).
Conflict of interestAll authors have no conflict of interest to declare.



