






Introduction & aim. Faldaprevir is a potent once-daily (q.d.) hepatitis C virus (HCV) NS3/4A protease inhibitor. The STARTVerso1 and STARTVerso2 phase 3 studies evaluated faldaprevir plus peginterferon alfa-2a/ribavirin (PegIFN/RBV) in treatment-naïve patients with chronic HCV genotype-1 infection.
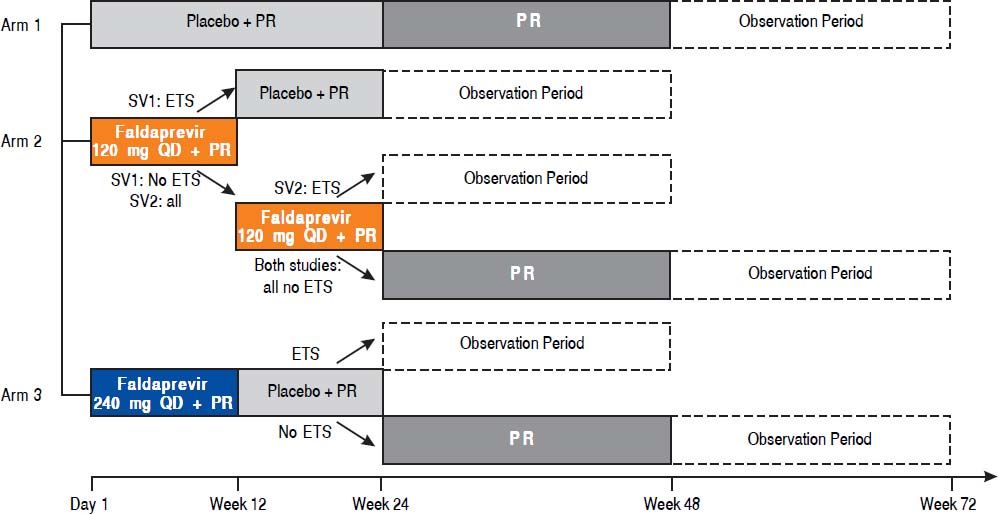
Material and methods. Patients were randomized 1:2:2 to receive placebo, faldaprevir 120 mg q.d. (12 or 24 weeks) or faldaprevir 240 mg q.d. (12 weeks) all with PegIFN/RBV (24-48 weeks). Faldaprevir 120 mg for 12 weeks only (STARTVerso1 only) required early treatment success (ETS, HCV RNA < 25 IU/mL at week 4 and undetected at week 8). All faldaprevir-treated patients with ETS stopped PegIFN/RBV at week 24. Primary endpoint: sustained virologic response 12 weeks post-treatment (SVR12).
Results. SVR12 rates were significantly higher for patients treated with faldaprevir 120 or 240 mg (72% and 73%, respectively) compared with placebo (50%); estimated differences (adjusted for trial, race, and genotype-1 subtype) faldaprevir 120 mg 24% (95% CI: 17-31%, P < 0.0001), faldaprevir 240 mg 23% (95% CI: 16-30%, P < 0.0001). Subgroup analyses consistently showed higher SVR12 rates for patients receiving faldaprevir compared with placebo. The incidence of adverse events (AEs) was similar in faldaprevir 120-mg and placebo groups and slightly higher in the faldaprevir 240-mg group. Serious AEs were reported in 6%, 7%, and 8% of patients in placebo, faldaprevir 120-mg, and faldaprevir 240-mg groups, respectively.
Conclusion. Addition of faldaprevir to PegIFN/RBV increased SVR12 in patients with HCV genotype-1, and was well tolerated. Faldaprevir 120 mg is effective in the treatment of HCV genotype-1. ClinicalTrials.gov: NCT01343888 and NCT01297270.
Chronic hepatitis C virus (HCV) infection affects more than 180 million people worldwide and more than 350,000 die each year as a result of related liver diseases, such as cirrhosis and hepatocellular carcinoma.1,2 Globally the most prevalent HCV genotype (GT) is GT-1.3 It is also the most difficult-to-treat GT with peginterferon alfa-2a and ribavirin (PegIFN/RBV).4,5 The introduction of direct-acting antiviral agents that can be used in combination regimens with or without PegIFN/RBV has represented a significant therapeutic advance in the treatment of HCV.6–8
Faldaprevir is a potent HCV NS3/4A protease inhibitor (PI) administered once daily (q.d.) as a single tablet, which has demonstrated efficacy in combination with PegIFN/ RBV in phase 2 and 3 studies.9–16 In treatment-naïve patients infected with HCV GT-1, sustained virologic response (SVR) rates of up to 84% were achieved with faldaprevir plus PegIFN/RBV.11,15 In addition, a phase 2 trial exploring duration (12 or 24 weeks) of faldaprevir 120 mg suggested no meaningful benefit of extending faldaprevir treatment from 12 to 24 weeks.13
The STARTVerso1 and STARTVerso2 phase 3 studies assessed the efficacy and safety of faldaprevir plus PegIFN/RBV in treatment-naïve patients with chronic HCV GT-1 infection in different geographic populations - Europe and Asia in STARTVerso1 and North America and Asia in STARTVerso2. The results of STARTVerso1 have been previously described.15 Here, we describe a preplanned pooled analysis of these two studies to allow further analysis of the efficacy and safety of faldaprevir in a larger dataset of treatment-naïve patients and for regional comparisons.
Material and MethodsStudy design and patientsSTARTVerso1 (NCT01343888) and STARTVerso2 (NCT01297270) were multicenter, randomized, doubleblind, placebo-controlled, parallel-group phase 3 studies. Patients were enrolled in Europe and Japan (STARTVer-so1) and in the USA, Canada, South Korea, and Taiwan (STARTVerso2). Eligible patients were treatment-naïve, aged 20-70 years (Japan) or 18–70 years (all other countries) and were chronically infected with HCV GT-1 diagnosed by positive anti-HCV antibodies and HCV RNA ≥ 1,000 IU/mL at screening in addition to a positive antibody or HCV RNA test > 6 months before screening, or a liver biopsy consistent with chronic HCV infection. Patients with compensated cirrhosis were eligible for inclusion. All patients had a liver biopsy within 3 years or a transient elastography (FibroScan®, Echosens, Paris, France) within 6 months of randomization to determine the stage of fibrosis (biopsies were not repeated if cirrhosis had been demonstrated more than 3 years prior to screening). For patients without a liver biopsy, fibrosis stage was classified on the basis of FibroScan results: < F3 for < 9.5 kPa, ≥ F3 for ≥ 9.5 kPa.17,18 Patients indicated to have cirrhosis but without biopsy or FibroScan were classified as having ≥ F3 fibrosis. The FibroScan threshold selected for cirrhosis was ≥ 13 kPa.19,20 Main exclusion criteria included mixed-GT HCV, human immunodeficiency virus or hepatitis B co-infection, decompensated liver disease, hemoglobin < 12 g/dL (women) or < 13 g/ dL (men), white blood cell count < 2,000 cells/mm3, absolute neutrophil count < 1,500 cells/mm3, and platelet count < 90,000 cells/mm3.
The documentation for each study, including protocol amendments, was approved by the appropriate institutional review board and the studies were carried out in accordance with the Declaration of Helsinki and International Conference on Harmonisation guidelines. All patients provided written informed consent before enrollment.
Independent data monitoring committees reviewed the efficacy and safety data at regular intervals during each study. A separate committee of dermatology experts was responsible for adjudicating cases of photosensitivity or rash of at least moderate intensity.
TreatmentsUsing an interactive voice response system, patients were randomized (1:2:2) to receive placebo for 24 weeks; faldaprevir 120 mg q.d. for 12 or 24 weeks; or faldaprevir 240 mg q.d. for 12 weeks, all in combination with PegIFN/RBV for 24 or 48 weeks (Supplementary figure 1). Randomization was stratified according to race (Black, Asian, other; with a 20% limit on patients from Asian countries) and HCV GT (GT-1a, GT-1b, other). PegIFN/ RBV plus placebo was chosen as a comparator because tel-aprevir and boceprevir became available in the USA only during the enrollment period and in other countries after completion of enrollment. Investigators, sponsors, and patients were blinded to treatment group up to week 24. HCV RNA results were blinded up to week 8.
Treatment duration in the faldaprevir groups was determined according to response, using early treatment success (ETS), defined as HCV RNA below the limit of quantification (25 IU/mL) at week 4 (target detected or target not detected [TND]) and TND at week 8. In STARTVerso1, patients received faldaprevir 120 mg for 12 weeks if they achieved ETS and for 24 weeks if they did not achieve ETS. In STARTVerso2, all patients in the faldaprevir 120 mg group received faldaprevir for 24 weeks. All faldaprevir-treated patients with ETS in both studies were directed to stop PegIFN/RBV at week 24.
For both studies, a loading dose of faldaprevir was administered on the first day (240 mg for the 120 mg group and 480 mg for the 240 mg group). Faldaprevir was administered once daily, in the morning. PegIFN (alfa-2a) 180 µg was administered subcutaneously once weekly. RBV was administered orally, twice daily at a total daily dose of 1,000 mg (for patients with body weight < 75 kg) or 1,200 mg (body weight ≥ 75 kg) except in Japan where the dose was 600 mg daily (body weight ≤ 60 kg), 800 mg daily (body weight > 60 and ≤ 80 kg), or 1,000 mg daily (body weight > 80 kg) in accordance with the local label. Both faldaprevir and RBV were given with food. Dose reductions were permitted for PegIFN/RBV (following approved medication guides), and dose interruptions were permitted for all three drugs if medically necessary. Fald-aprevir monotherapy was not permitted.
All study medication was stopped in the event of: viro-logic rebound at or after week 4 (increase in HCV RNA ≥ 1 log10 from nadir or > 25 IU/mL after an initial decrease to < 25 IU/mL); or lack of early virologic response (decrease in HCV RNA ≥ 2 log10 from baseline at week 12); or lack of virologic response (HCV RNA > 25 IU/mL at week 24).
Outcome measuresThe primary endpoint for each study was SVR12 weeks post-treatment (SVR12; HCV RNA < 25 IU/mL TND 12 weeks after planned completion of therapy). Secondary efficacy endpoints included: ETS (HCV RNA < 25 IU/mL at week 4 and < 25 IU/mL TND at week 8), rapid viro-logic response (RVR; HCV RNA < 25 IU/mL at week 4), complete early virologic response (cEVR; HCV RNA < 25 IU/mL TND at week 12), end of treatment response (ETR; HCV RNA < 25 IU/mL TND at end of all treatment), and SVR24 weeks post-treatment (SVR24; HCV RNA < 25 IU/mL TND 24 weeks after completion of therapy). Plasma HCV RNA levels were measured using the Roche COBAS®Taqman HCV/HPS assay (Roche Diagnostics Limited, Burgess Hill, West Sussex, UK).
Resistance was assessed by population-based sequencing of the HCV NS3/4A region (amino acids 1–685) performed at Janssen Diagnostics (Beerse, Belgium) with lower limit of variant detection of 30%. HCV NS3/4A sequence was derived from baseline plasma samples from all patients and from post-baseline samples including post-treatment follow-up from patients who did not achieve SVR.
Safety was assessed by monitoring adverse events (AEs) and laboratory parameters throughout the trial and for 28 days after the end of treatment. With the exception of photosensitivity, these were graded using the Division of AIDS Table for Grading the Severity of Adult and Pediat-ric Adverse Events. Photosensitivity was graded as mild (grade 1), moderate (grade 2), or severe (grade 3) according to predefined criteria. Rash or photosensitivity of at least moderate intensity were designated as AEs of special interest and were assessed by an independent rash adjudication committee. A rash management plan was used to minimize the risk of rash and photosensitivity and patients were advised to restrict exposure to sun and artificial UV light and to use sun-block SPF 30 or higher.
Statistical analysisPooling of data from the two studies to achieve a larger data set was justified since the study designs and the results were similar with respect to comparison to placebo. The integrated efficacy analysis population included all randomized patients who received at least one dose of study medication. The integrated safety analysis set included all patients who received at least one dose of study medication regardless of randomization.
The proportions of patients with SVR12 in the pooled population were analyzed using the Cochran-Mantel-Haenszel test, stratified by trial, race, and GT-1 subtype. For comparison between faldaprevir dose groups, confidence intervals for differences in SVR12 rates were determined using Greenland’s formula with continuity correction (Koch’s method) and compared with a non-inferiority limit pre-defined at 12%. All other efficacy and safety data were summarized descriptively.
A stepwise multiple logistic regression analysis was used to evaluate the effect of covariates (age, sex, race, body mass index, presence of cirrhosis, baseline gamma-glutamyl-transferase (GGT), baseline alanine aminotransferase (ALT), HCV subtype, IL28B genotype, treatment, and baseline HCV RNA levels) on virologic response rates.
Protocol amendmentsIn accordance with updated scientific knowledge, for both studies, the definition of SVR was changed in protocol amendments from SVR24 to SVR12.21 Other protocol amendments included implementation of the Division of AIDS Table for Grading the Severity of Adult and Pediat-ric Adverse Events.
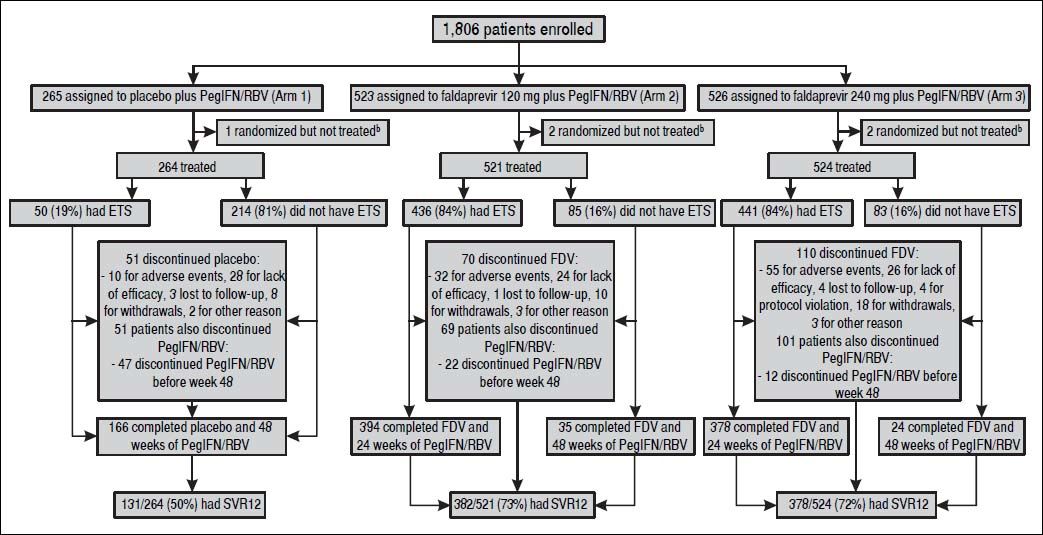
ResultsPatient populationPatients were recruited from April 2011 to October 2011 in 15 countries. The last patient completed the SVR12 visit in February 2013. A total of 1309 patients were randomized and treated across STARTVerso1 and STARTVerso2 (Figure 1).
 at week 4 and TND at week 8. PegIFN/RBV: pegylated interferon alfa-2a/ribavirin. SVR12: sustained virologic response HCV RNA < 25 IU/mL (TND) 12 weeks after completion of therapy. TND: target not detected. a A total of 492 patients failed screening due to: adverse events (1), inclusion/exclusion criteria not met (401), lost to follow-up (8), withdrawal (53), and other reasons (29). b Five patients were randomized but not treated due to protocol violations (2 patients from arm 2 and 1 patient from arm 3) and other reasons (1 patient from arm 1 and 1 patient from arm 3).")
Patient flow/disposition a. ETS: early treatment success, defined as HCV RNA < 25 IU/mL (target detected or TND) at week 4 and TND at week 8. PegIFN/RBV: pegylated interferon alfa-2a/ribavirin. SVR12: sustained virologic response HCV RNA < 25 IU/mL (TND) 12 weeks after completion of therapy. TND: target not detected. a A total of 492 patients failed screening due to: adverse events (1), inclusion/exclusion criteria not met (401), lost to follow-up (8), withdrawal (53), and other reasons (29). b Five patients were randomized but not treated due to protocol violations (2 patients from arm 2 and 1 patient from arm 3) and other reasons (1 patient from arm 1 and 1 patient from arm 3).
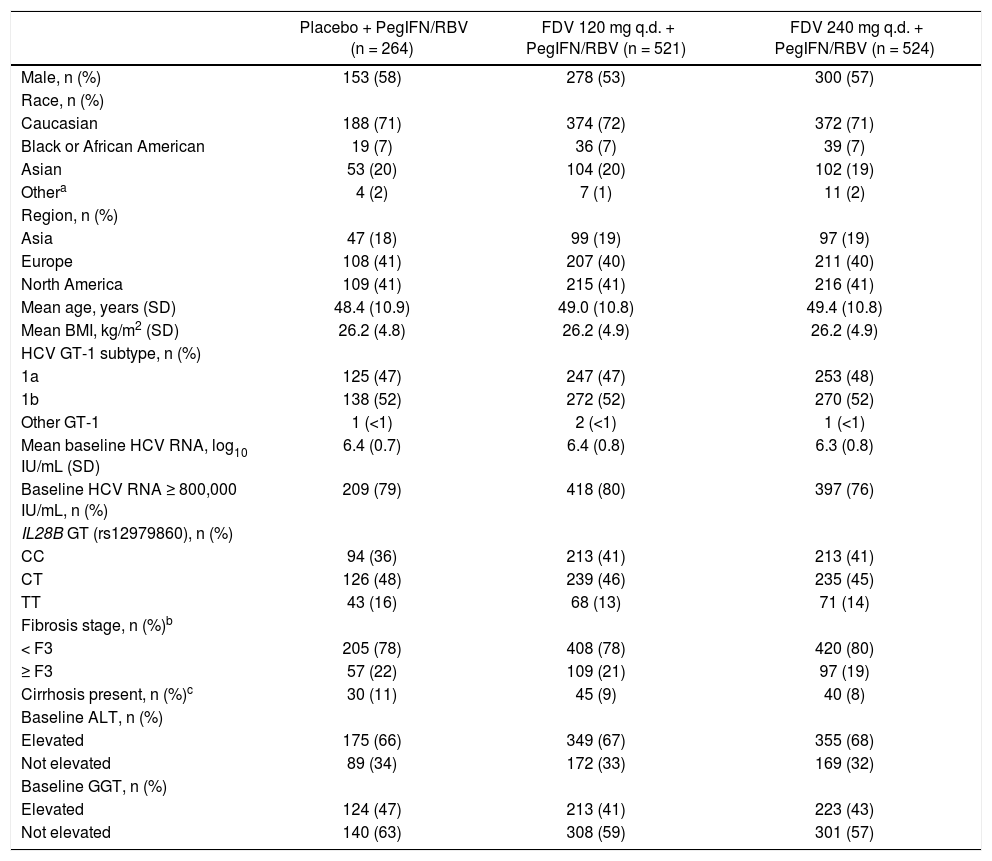
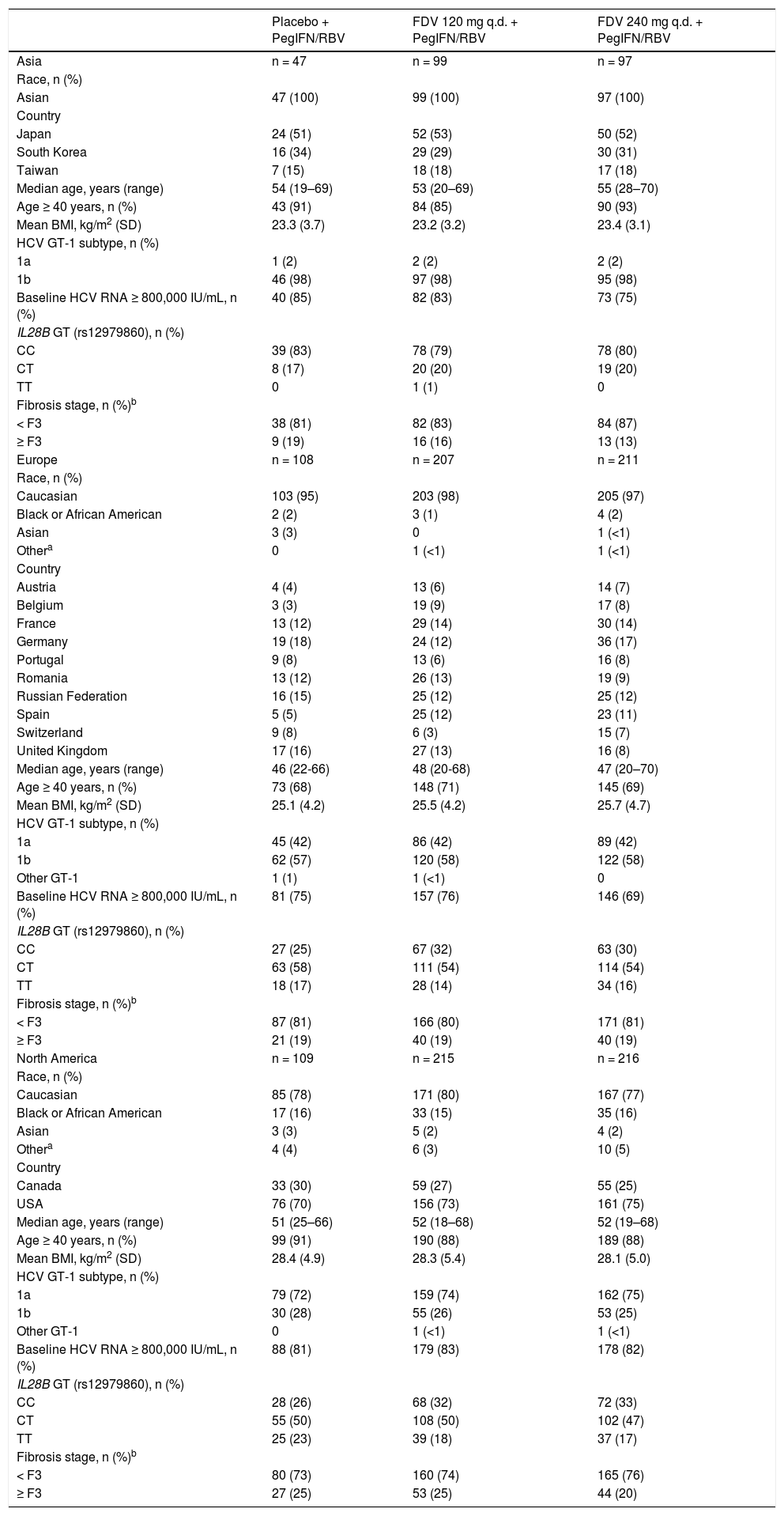
Baseline patient demographics and disease characteristics were well balanced across the pooled treatment groups (Table 1). Just under half of the patients were infected with HCV GT-1a across the two studies. Among patients with GT-1a infection, the NS3 Q80K variant was detected alone or in a mixture with other variants in baseline plasma samples from 213/616 (35%) patients. Baseline patient demographics and disease characteristics by region are provided in the supplementary information (Supplementary table 1). In North America there were more Black/African American patients (16 vs. 2% in Europe and none in Asia) and more patients with a higher mean body mass index (approximately 28 kg/m2vs. approximately 25 kg/m2 in Europe and 23 kg/m2 in Asia). Patients in the European study population tended to be younger than those in Asia and North America (proportions ≥ 40 years 70 vs. 89% and 89%, respectively). Overall, approximately 40% of patients were IL28B (rs12979860) CC genotype with a greater proportion in Asia (80%) than in Europe and North America (30 and 31%, respectively). HCV GT-1b subtype was recorded in 98% of patients in Asia compared with 58 and 26% in Europe and North America, respectively. In North America there were higher proportions of patients with baseline HCV RNA of ≥ 800,000 IU/mL (82 vs. 73% in Europe and 80% in Asia), patients with HCV GT-1a (74 vs. 42% in Europe and 2% in Asia), patients with ≥ F3 fi-brosis (23 vs. 19% in Europe and 16% in Asia), or with compensated cirrhosis (determined by the investigator: 11 vs. 7% in both Europe and Asia). Metavir fibrosis stage was most frequently used to assess liver fibrosis in North America and Asia (88 and 63%, respectively, vs. Europe 38%) while FibroScan was most frequently used in Europe (65 vs. 11% in North America and 38% in Asia).
Baseline demographics and disease characteristics.
| Placebo + PegIFN/RBV (n = 264) | FDV 120 mg q.d. + PegIFN/RBV (n = 521) | FDV 240 mg q.d. + PegIFN/RBV (n = 524) | |
|---|---|---|---|
| Male, n (%) | 153 (58) | 278 (53) | 300 (57) |
| Race, n (%) | |||
| Caucasian | 188 (71) | 374 (72) | 372 (71) |
| Black or African American | 19 (7) | 36 (7) | 39 (7) |
| Asian | 53 (20) | 104 (20) | 102 (19) |
| Othera | 4 (2) | 7 (1) | 11 (2) |
| Region, n (%) | |||
| Asia | 47 (18) | 99 (19) | 97 (19) |
| Europe | 108 (41) | 207 (40) | 211 (40) |
| North America | 109 (41) | 215 (41) | 216 (41) |
| Mean age, years (SD) | 48.4 (10.9) | 49.0 (10.8) | 49.4 (10.8) |
| Mean BMI, kg/m2 (SD) | 26.2 (4.8) | 26.2 (4.9) | 26.2 (4.9) |
| HCV GT-1 subtype, n (%) | |||
| 1a | 125 (47) | 247 (47) | 253 (48) |
| 1b | 138 (52) | 272 (52) | 270 (52) |
| Other GT-1 | 1 (<1) | 2 (<1) | 1 (<1) |
| Mean baseline HCV RNA, log10 IU/mL (SD) | 6.4 (0.7) | 6.4 (0.8) | 6.3 (0.8) |
| Baseline HCV RNA ≥ 800,000 IU/mL, n (%) | 209 (79) | 418 (80) | 397 (76) |
| IL28B GT (rs12979860), n (%) | |||
| CC | 94 (36) | 213 (41) | 213 (41) |
| CT | 126 (48) | 239 (46) | 235 (45) |
| TT | 43 (16) | 68 (13) | 71 (14) |
| Fibrosis stage, n (%)b | |||
| < F3 | 205 (78) | 408 (78) | 420 (80) |
| ≥ F3 | 57 (22) | 109 (21) | 97 (19) |
| Cirrhosis present, n (%)c | 30 (11) | 45 (9) | 40 (8) |
| Baseline ALT, n (%) | |||
| Elevated | 175 (66) | 349 (67) | 355 (68) |
| Not elevated | 89 (34) | 172 (33) | 169 (32) |
| Baseline GGT, n (%) | |||
| Elevated | 124 (47) | 213 (41) | 223 (43) |
| Not elevated | 140 (63) | 308 (59) | 301 (57) |
FibroScan® results were used to determine stage of fibrosis for patients without a liver biopsy [< F3 = < 9.5 kPa, ≥ F3 = ≥ 9.5 kPa). If a patient was indicated to have cirrhosis but had neither biopsy nor FibroScan data they were classified as having ≥ F3 fibrosis.
Cirrhosis was determined by the investigator based on FibroScan, biopsy, and/or other clinical parameters. ALT: alanine aminotransferase. BMI: body mass index. FDV: faldaprevir. GGT: gamma-glutamyltranspeptidase. GT: genotype. HCV: hepatitis C virus. PegIFN/RBV: peginterferon alfa-2a/ribavirin. q.d.: once daily. SD: standard deviation.
SVR12 rates were significantly higher for patients treated with faldaprevir 120 or 240 mg (73 and 72%, respectively) compared with placebo (50%) (Table 2). Adjusted for trial, race, and HCV GT-1 subtype, the estimated differences in SVR12 of faldaprevir vs. placebo were 24% (95% confidence interval [CI]: 17–31) for faldaprevir 120 mg (P < 0.0001) and 23% (95% CI: 16–30) for faldaprevir 240 mg (P < 0.0001). No difference was observed in the proportion of patients who achieved SVR12 in the faldaprevir 120 mg group compared with the 240 mg group (estimated difference in SVR12 0.6% [95% CI: -4-6]; P < 0.0001 for 12% non-inferiority). In each of the faldaprevir dose groups, ETS was achieved by 84% of patients and 83% of these patients achieved SVR12 (Table 2). Rates of other vi-rologic endpoints (RVR, cEVR, and ETR) did not differ between the faldaprevir dose groups and were consistently higher in both groups compared with the placebo group (Table 2).
Response according to treatment group (full analysis set).
| Placebo + PegIFN/RBV (n = 264) | FDV 120 mg q.d. + PegIFN/RBV (n = 521) | FDV 240 mg q.d. + PegIFN/RBV (n = 524) | |
|---|---|---|---|
| SVR12, n (%) | 131 (50) | 382 (73) | 378 (72) |
| Estimated Δ vs. placeboa (95% CI) | 24% (17–31) | 23% (16–30) | |
| P-value | P < 0.0001 | P < 0.0001 | |
| Estimated Δ vs. FDV 240 mgb (95% CI) | 0.6% (-4–6) | ||
| P-value | P < 0.0001 | ||
| RVR, n (%) | 51 (19) | 459 (88) | 472 (90) |
| ETS, n (%) | 50 (19) | 436 (84) | 441 (84) |
| ETS and SVR12, n/N (%) | 41/50 (82) | 362/436 (83) | 368/441 (83) |
| cEVR, n (%) | 120 (45) | 452 (87) | 443 (85) |
| ETR, n (%) | 189 (72) | 467 (90) | 461 (88) |
CI: confidence interval. ETR: end of treatment response (HCV RNA < 25 IU/mL [TND] at end of all treatment). ETS: early treatment success (HCV RNA < 25 IU/mL [target detected or TND] at week 4 and TND at week 8). FDV: faldaprevir. PegIFN/RBV: peginterferon alfa-2a/ribavirin. q.d.: once daily. RVR: rapid virologic response (HCV RNA < 25 IU/mL [target detected or TND] at week 4). SVR12: sustained virologic response (HCV RNA < 25 IU/mL [TND]) 12 weeks after completion of therapy. TND: target not detected.
A lower proportion of patients experienced a null or partial response in the faldaprevir groups (1% in each group) than in the placebo group (14%). On-treatment breakthrough occurred in 7% of patients in each of the faldaprevir dose groups and in 8% of patients in the placebo group (Table 3). Among patients who completed treatment and had an ETR, relapse was reported for 15 and 10% of patients in the faldaprevir 120 and 240 mg dose groups, respectively, and for 21% of the placebo group (Table 3).
Treatment failure.
| n (%) | Placebo + PegIFN/RBV (n = 264) | FDV 120 mg q.d. + PegIFN/RBV (n = 521) | FDV 240 mg q.d. + PegIFN/RBV (n = 524) |
|---|---|---|---|
| On treatment failure | 59 (22) | 41 (8) | 40 (8) |
| Breakthrougha | 21 (8) | 34 (7) | 37 (7) |
| ≤ Week 12 | 4 (2) | 15(3) | 16(3) |
| > Week 12 – ≤ Week 24 | 9(3) | 14(3) | 16(3) |
| > Week 24 | 8(3) | 5 (1) | 5 (1) |
| Null or partial responseb | 38 (14) | 7 (1) | 3 (1) |
| Relapsec | 33/160 (21) | 63/424 (15) | 41/393 (10) |
Confirmed HCV RNA ≥ 25 IU/mL after initial decrease to < 25 IU/mL (target detected or TND) or ≥ 1 log10 in those with nadir ≥ 25 IU/mL.
Among patients who did not achieve SVR12 and had available HCV NS3 sequence data, treatment-emergent resistance-associated variants (RAVs) at R155 and/or D168 codons were detected in GT-1a samples from 145/154 (94%) patients and GT-1b samples from 68/85 (80%) patients. The predominant treatment-emergent RAVs were R155K (88%; 136/154) in GT-1a, and D168 substitutions (72%; 61/85) in GT-1b (predominantly D168V 56%; 48/85). Among patients with on-treatment breakthrough on faldaprevir, 100% of sequences for both genotypes encoded R155 and/or D168 RAVs. Among patients with post-treatment relapse, a lack of detectable RAVs was more common for GT-1b (16%; 6/38) than for GT-1a (5%; 3/64).
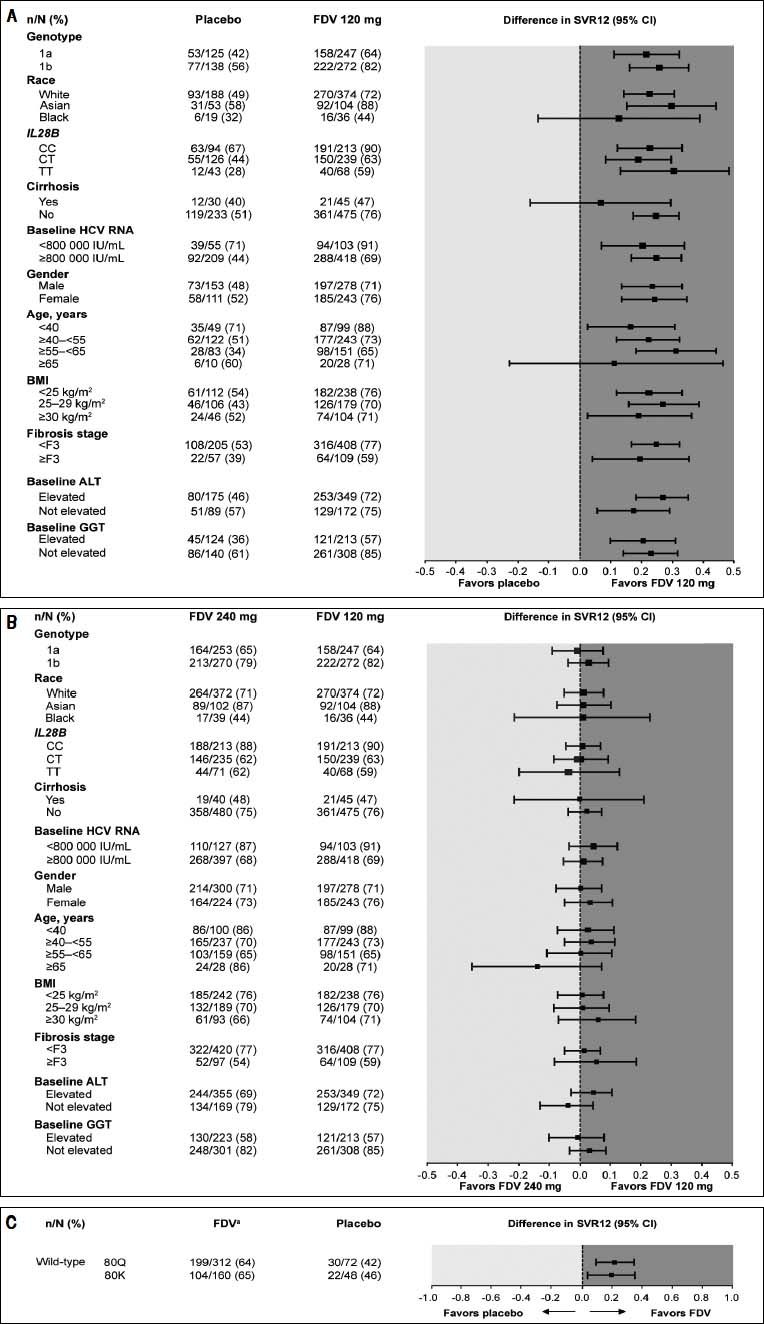
Analyses of SVR12 by subgroups consistently showed higher rates for patients treated with faldaprevir 120 or 240 mg compared with placebo, including among patients in whom the NS3 Q80K variant was detected at baseline (Figure 2). SVR12 rates in all treatment arms were highest in Asia and lowest in North America (Supplementary table 2). A breakdown of these data by country highlights that the lower SVR12 rates reported for North America are attributable to the US patient cohort where 61% (95/156) of patients treated with faldaprevir 120 mg, 55% (88/161) of patients treated with faldaprevir 240 mg, and 42% (32/76) of patients treated with placebo achieved SVR12. The lower SVR12 rates seen in the faldaprevir 120 mg and placebo arms are explained by differences in baseline characteristics, mainly HCV GT-1 subtype, degree of fibrosis, age, and HCV RNA. While these factors also played a role in the lower SVR12 rate observed in the faldaprevir 240 mg arm, analysis of reasons for failure to achieve SVR12 suggested higher rates of premature discontinuations for reasons other than virologic failure in North America than in Asia and Europe (9 vs. 6% and 4%, respectively) (Supplementary table 3). Investigations were carried out to look for possible reasons for increased discontinuation rates in North America but no clear pattern emerged.
 the placebo and faldaprevir 120 mg groups and B) faldaprevir 120 mg and 240 mg groups, and C) differences in SVR12 rates between pooled fald-aprevir treatment groups and placebo according to presence of GT-1a Q80K at baseline. ALT: alanine aminotrans-ferase. BMI: body mass index. CI: confidence interval. FDV: faldaprevir. GGT: gamma-glutamyltranspepti-dase. HCV: hepatitis C virus. SVR12: sustained virologic response 12 weeks after completion of therapy. aPooled FDV treatment groups.")
Forest plot showing differences in various patient subgroups between SVR12 rates in A) the placebo and faldaprevir 120 mg groups and B) faldaprevir 120 mg and 240 mg groups, and C) differences in SVR12 rates between pooled fald-aprevir treatment groups and placebo according to presence of GT-1a Q80K at baseline. ALT: alanine aminotrans-ferase. BMI: body mass index. CI: confidence interval. FDV: faldaprevir. GGT: gamma-glutamyltranspepti-dase. HCV: hepatitis C virus. SVR12: sustained virologic response 12 weeks after completion of therapy. aPooled FDV treatment groups.
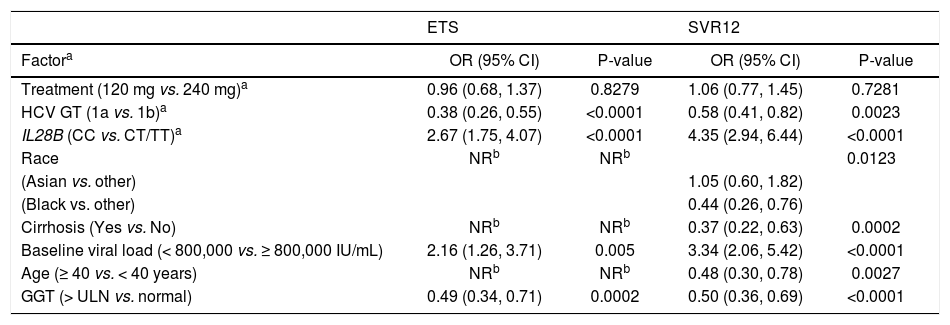
To explore the possible impact of differences in patient characteristics on rates of SVR12, a multiple logistic regression analysis was carried out. HCV GT-1 subtype, IL28B genotype, baseline HCV RNA, and GGT were found to be significantly (P < 0.05) associated with both SVR12 and ETS (Table 4). Race, cirrhosis, and age were significant predictors of SVR12 but not ETS. Faldaprevir dose was not associated with ETS or SVR12.
Stepwise logistic regression analysis of baseline factors predictive of virologic response in faldaprevir-treated patients.
| ETS | SVR12 | |||
|---|---|---|---|---|
| Factora | OR (95% CI) | P-value | OR (95% CI) | P-value |
| Treatment (120 mg vs. 240 mg)a | 0.96 (0.68, 1.37) | 0.8279 | 1.06 (0.77, 1.45) | 0.7281 |
| HCV GT (1a vs. 1b)a | 0.38 (0.26, 0.55) | <0.0001 | 0.58 (0.41, 0.82) | 0.0023 |
| IL28B (CC vs. CT/TT)a | 2.67 (1.75, 4.07) | <0.0001 | 4.35 (2.94, 6.44) | <0.0001 |
| Race | NRb | NRb | 0.0123 | |
| (Asian vs. other) | 1.05 (0.60, 1.82) | |||
| (Black vs. other) | 0.44 (0.26, 0.76) | |||
| Cirrhosis (Yes vs. No) | NRb | NRb | 0.37 (0.22, 0.63) | 0.0002 |
| Baseline viral load (< 800,000 vs. ≥ 800,000 IU/mL) | 2.16 (1.26, 3.71) | 0.005 | 3.34 (2.06, 5.42) | <0.0001 |
| Age (≥ 40 vs. < 40 years) | NRb | NRb | 0.48 (0.30, 0.78) | 0.0027 |
| GGT (> ULN vs. normal) | 0.49 (0.34, 0.71) | 0.0002 | 0.50 (0.36, 0.69) | <0.0001 |
TND: target not detected. ULN: upper limit of normal.
aTreatment, GT-1 subtype, and IL28B were fixed to stay in the model. Other variables included for selection were: race (Black, Asian, other), cirrhosis (yes, no), baseline viral load (< 800,000, ≥ 800,000 IU/mL), baseline GGT (normal, elevated), baseline alanine aminotransferase (normal, elevated), sex (male, female), age (< 40, ≥ 40 years), body mass index (< 25, ≥ 25 kg/m2), and study (STARTVerso1, STARTVerso2). Only those retained in the model after stepwise selection (P < 0.05) are included in the table.
bNot retained in the model after stepwise selection (P < 0.05). CI: confidence interval. ETS: early treatment success (HCV RNA < 25 IU/mL [target detected or TND] at week 4 and TND at week 8. GGT: gamma-gliutamyltranspeptidase. GT: genotype. HCV: hepatitis C virus. NR: not retained. OR: odds ratio. SVR12: sustained virologic response (HCV RNA < 25 IU/mL [TND]) 12 weeks after completion of therapy.
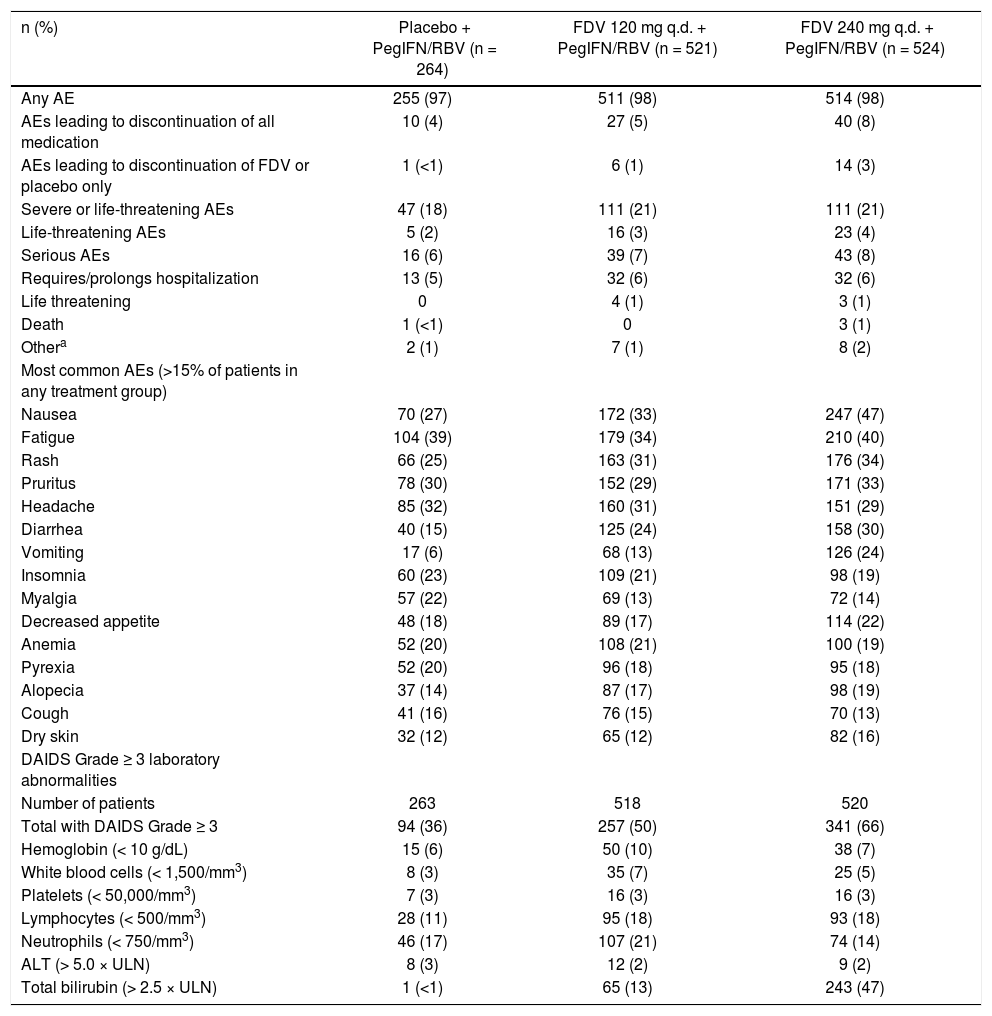
The incidence of any AEs, including AEs leading to discontinuation of all medication or of faldaprevir/place-bo, was similar across the placebo and faldaprevir 120 mg treatment groups and slightly higher for the faldaprevir 240 mg group (Table 5). A slightly higher proportion of patients in STARTVerso2 compared with STARTVerso1 had AEs leading to discontinuation of all medication in the faldaprevir 120 mg (6 vs. 4%, respectively) and 240 mg (10 vs. 5%, respectively) treatment groups. Serious AEs were reported in 6, 7, and 8% of patients in the placebo, faldaprevir 120 mg, and faldaprevir 240 mg groups, respectively.
Summary of adverse events and laboratory abnormalities.
| n (%) | Placebo + PegIFN/RBV (n = 264) | FDV 120 mg q.d. + PegIFN/RBV (n = 521) | FDV 240 mg q.d. + PegIFN/RBV (n = 524) |
|---|---|---|---|
| Any AE | 255 (97) | 511 (98) | 514 (98) |
| AEs leading to discontinuation of all medication | 10 (4) | 27 (5) | 40 (8) |
| AEs leading to discontinuation of FDV or placebo only | 1 (<1) | 6 (1) | 14 (3) |
| Severe or life-threatening AEs | 47 (18) | 111 (21) | 111 (21) |
| Life-threatening AEs | 5 (2) | 16 (3) | 23 (4) |
| Serious AEs | 16 (6) | 39 (7) | 43 (8) |
| Requires/prolongs hospitalization | 13 (5) | 32 (6) | 32 (6) |
| Life threatening | 0 | 4 (1) | 3 (1) |
| Death | 1 (<1) | 0 | 3 (1) |
| Othera | 2 (1) | 7 (1) | 8 (2) |
| Most common AEs (>15% of patients in any treatment group) | |||
| Nausea | 70 (27) | 172 (33) | 247 (47) |
| Fatigue | 104 (39) | 179 (34) | 210 (40) |
| Rash | 66 (25) | 163 (31) | 176 (34) |
| Pruritus | 78 (30) | 152 (29) | 171 (33) |
| Headache | 85 (32) | 160 (31) | 151 (29) |
| Diarrhea | 40 (15) | 125 (24) | 158 (30) |
| Vomiting | 17 (6) | 68 (13) | 126 (24) |
| Insomnia | 60 (23) | 109 (21) | 98 (19) |
| Myalgia | 57 (22) | 69 (13) | 72 (14) |
| Decreased appetite | 48 (18) | 89 (17) | 114 (22) |
| Anemia | 52 (20) | 108 (21) | 100 (19) |
| Pyrexia | 52 (20) | 96 (18) | 95 (18) |
| Alopecia | 37 (14) | 87 (17) | 98 (19) |
| Cough | 41 (16) | 76 (15) | 70 (13) |
| Dry skin | 32 (12) | 65 (12) | 82 (16) |
| DAIDS Grade ≥ 3 laboratory abnormalities | |||
| Number of patients | 263 | 518 | 520 |
| Total with DAIDS Grade ≥ 3 | 94 (36) | 257 (50) | 341 (66) |
| Hemoglobin (< 10 g/dL) | 15 (6) | 50 (10) | 38 (7) |
| White blood cells (< 1,500/mm3) | 8 (3) | 35 (7) | 25 (5) |
| Platelets (< 50,000/mm3) | 7 (3) | 16 (3) | 16 (3) |
| Lymphocytes (< 500/mm3) | 28 (11) | 95 (18) | 93 (18) |
| Neutrophils (< 750/mm3) | 46 (17) | 107 (21) | 74 (14) |
| ALT (> 5.0 × ULN) | 8 (3) | 12 (2) | 9 (2) |
| Total bilirubin (> 2.5 × ULN) | 1 (<1) | 65 (13) | 243 (47) |
Other serious AEs include those that persist or cause significant disability/incapacity, or other medically important serious events. AE: adverse event. ALT: alanine aminotransferase. DAIDS: Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events. FDV: faldaprevir. PegIFN/RBV: peginterferon alfa-2a/ribavirin. q.d.: once daily. ULN: upper limit of normal.
Common AEs were those associated with PegIFN/ RBV, with the exception of rash and gastrointestinal AEs, which were reported by a greater proportion of faldapre-vir-treated patients compared with those who received placebo. The incidence of these AEs was generally higher in the 240 mg arm than the 120 mg arm (Table 5). Rash was mainly mild (87% of rashes on placebo and 79% of rashes on faldaprevir) and there were no cases of life-threatening rash. Discontinuation of any study medication due to rash was 1% in each treatment group. Photosensitivity was infrequent: 2% of patients treated with placebo, 1% of patients treated with faldaprevir 120 mg, and 4% of patients treated with faldaprevir 240 mg. All cases were mild except for three cases of moderate intensity photosensitivity in the faldaprevir 240 mg group. No patients experienced photosensitivity as a serious AE and no patients discontinued due to photosensitivity.
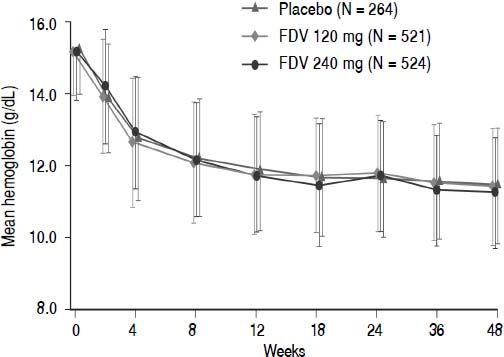
Abnormalities in hemoglobin, white blood cells, platelets, neutrophils, and ALT occurred with similar frequency across the placebo and faldaprevir treatment groups (Table 5). There were no differences between the treatment groups in terms of hemoglobin reductions over time (Figure 3). Total bilirubin levels > 2.5 times the upper limit of normal were more frequent in the faldaprevir groups, were mainly due to an increase in unconjugated bilirubin, and were not associated with increased levels of ALT or aspartate transaminase (AST). Bilirubin elevations were characterized by a predominance of unconjugated bilirubin, peaked around week 2, reached a plateau from week 2 to end of treatment, and rapidly returned to baseline levels in all patients shortly after completion of fald-aprevir treatment. Jaundice was reported as an AE in 4% of patients receiving faldaprevir 120 mg and 14% of patients receiving faldaprevir 240 mg compared with 0.4% of patients in the placebo group. Discontinuation of faldaprevir or placebo due to jaundice was reported in 1 patient (0.2%) treated with faldaprevir 120 mg and 5 patients (1%) treated with faldaprevir 240 mg. In 3 of the patients (0.6%) in the faldaprevir 240 mg treatment group, jaundice led to discontinuation of all study medication.
Discussion
This pooled analysis of data from the STARTVerso1 and 2 studies shows that once-daily faldaprevir combined with PegIFN/RBV is efficacious in treatment-naïve patients with HCV GT-1. A large proportion (84%) of patients who received faldaprevir plus PegIFN/RBV were able to stop all treatment at Week 24, of whom 83% achieved SVR12. Efficacy was comparable across faldaprevir dose (120 or 240 mg), treatment duration (12 or 24 weeks), and across subgroups analyzed. Furthermore, the addition of faldaprevir 120 mg to PegIFN/RBV resulted in a tolera-bility profile similar to PegIFN/RBV alone.
The pooled data demonstrate that SVR12 rates were significantly higher in both the faldaprevir 120 and 240 mg treatment arms (73 and 72%, respectively) compared with PegIFN/RBV alone (50%). The similar efficacy between the 120 mg and 240 mg dose groups was evident in both trials across all subgroups analyzed. Multivariate analysis identified baseline predictors of SVR that are typically associated with response to PegIFN/RBV (with or without direct-acting antivirals), such as IL28B genotype, baseline HCV RNA level, and cirrhosis.22–24 HCV GT-1 subtype b was also a predictor of response, although treatment differences in SVR rates between faldaprevir and PegIFN/ RBV alone were similar within GT-1a and -1b subgroups. Reduced response rates in patients with HCV GT-1a have also been reported with other HCV PIs,24,25 and are likely related to the lower barrier to resistance presented by this subtype.26–28 Of note, the presence of the Q80K variant, which has been shown to compromise the efficacy of the PI simeprevir,25 had no impact on SVR rates in these studies, confirming the lack of impact of this substitution on response to faldaprevir reported in phase 2 studies.26 HCV NS3 resistance-associated variants were detected in the majority of virologic failures; predominant changes in GT-1a (R155K) and GT-1b (D168V) were consistent with previous reports.26–28
SVR12 varied according to recruitment region, with rates lowest among patients from study centers in North America, specifically the USA, compared with patients from study centers in Europe and Asia. These regional differences were likely influenced by differences in baseline factors. Compared with Europe and Asia, the North America patient population included a higher proportion of patients with characteristics that were negative prognostic factors for SVR12 such as HCV GT-1a, ≥ F3 fibrosis, age ≥ 40 years, and baseline HCV RNA ≥ 800,000 IU/ mL. There were also more premature discontinuations for reasons other than virologic failure (e.g. AEs and withdrawal of consent) in North American subjects compared with Europe. Other factors may also have had an influence; however, patient numbers were not high enough to reliably investigate all possible drivers of the observed difference in response rate. It should be noted that, regardless of regional and subgroup differences in absolute SVR12 rates, the response observed in faldaprevir treatment groups remained consistently superior to the response with PegIFN/RBV alone.
Faldaprevir in combination with PegIFN/RBV was generally well tolerated, with the most frequent AEs associated with PegIFN/RBV treatment. Treatment discontinuation due to AEs was similar in the 120 mg and placebo groups (4 and 5%, respectively) and slightly more common with faldaprevir 240 mg (8%). Reductions in hemoglobin were similar between the placebo and faldaprevir study arms, and the incidence of anemia was not increased with faldaprevir plus PegIFN/RBV compared with PegIFN/RBV alone. In contrast, the rate of anemia increased when telaprevir or boceprevir were combined with PegIFN/RBV, and severe anemia was reported as a common AE in real-world studies of these first-generation PIs.29 The addition of faldaprevir to PegIFN/RBV resulted in dose-dependent increases in the incidence of rash, gastrointestinal events, and hyperbilirubinemia, consistent with results of phase 2 studies.11,12 The majority of rash events were mild in intensity, and led to discontinuation in only 1% of patients who received faldaprevir. Pho-tosensitivity events were rare, likely as a result of the sun-protection measures recommended as part of this trial. The increase in bilirubin in the STARTVerso1 and 2 trials was transient and benign, and predominantly the result of elevations in unconjugated bilirubin. Faldaprevir reversibly inhibits the bilirubin conjugation enzyme UGT1A1, and to a lesser extent the bilirubin transporters OATP1B1 and MRP2.30 Reversible increases in bilirubin have also been observed with simeprevir, although primarily driven by inhibition of OATP1B1 and MRP2.31
A limitation of this pooled analysis is the absence of directly comparable 12- and 24-week faldaprevir treatment arms, as treatment duration varied across trials and early treatment response. However, pre-specified cross-dose and trial comparison suggests no effect of treatment duration with faldaprevir, which is supported by phase 2 data and phase 3 in HIV co-infected patients.13,32 Another possible limitation is that the potent antiviral activity of fald-aprevir to week 8, and/or the effects of this drug on bilirubin, could have been used to infer treatment allocation before unblinding. However, few patients withdrew consent in this study so the effect is likely to be minimal.
In conclusion, this pooled analysis of the STARTVerso1 and 2 data confirmed that the combination of faldaprevir and PegIFN/RBV significantly increased SVR12 rates compared with PegIFN/RBV alone in treatment-naïve patients with HCV GT-1 infection, and that faldaprevir 120 mg and 240 mg were equally efficacious. In this large dataset, responses with faldaprevir were consistently superior to those with PegIFN across subgroups analyzed, including regional groups, although achievement of SVR12 was influenced by baseline variables typically associated with PegIFN/RBV responsiveness. Across both studies fald-aprevir and PegIFN/RBV was well tolerated, although tol-erability was slightly better in the 120 mg treatment arm compared with the 240 mg arm. The addition of faldapre-vir 120 mg to PegIFN/RBV thus represents an efficacious regimen, with improved tolerability and convenience compared with first-generation PIs. Since the completion of this study, faldaprevir development has been terminated in view of the newly available interferon-free regimens demonstrating substantially higher response rates, better tolerability, and shorter treatment durations. The results presented here mark an important step in the progress of HCV research and demonstrate the commitment of patients and investigators to finding a cure for chronic hepatitis C. The results provide valuable information about HCV NS3 PI therapy in treatment-naïve patients that may be relevant to the use of other direct-acting antivirals across a broad range of global regions, including some where PegIFN/RBV may continue to be a component of first-line therapy.
Abbreviations- •
AE: adverse event.
- •
ALT: alanine aminotransferase.
- •
AST: aspartate transaminase.
- •
cEVR: complete early virologic response.
- •
CI: confidence interval.
- •
ETR: end of treatment response.
- •
ETS: early treatment success.
- •
GGT: gamma-glutamyltransferase.
- •
GT: genotype.
- •
HCV: hepatitis C virus.
- •
PegIFN/RBV: peginterferon alfa-2a and ribavirin.
- •
PI: protease inhibitor.
- •
q.d.: once daily.
- •
RAVs: resistance-associated variants.
- •
RVR: rapid virologic response.
- •
SPF: sun protection factor.
- •
SVR: sustained virologic response.
- •
SVR12: SVR 12 weeks post-treatment.
- •
SVR24: SVR 24 weeks post-treatment.
- •
TND: target not detected.
- •
UV: ultra-violet.
This study was funded by Boehringer Ingelheim Phar-ma GmbH & Co. KG. Writing support was provided by Esther Race of Choice Healthcare Solutions and funded by Boehringer Ingelheim Pharma GmbH & Co. KG.
Author ContributionsGuarantor of the article: JO Stern.DMJ, TA, DD, GRF, MSS, SZ, PM, EMY, CM, DO, MW, LEM, RB, MB, TH, SN, J-HK, MO, SWP, DKW, ET, KK, SVF, and PF were study investigators and contributed to recruitment of patients. DMJ, TA, DD, GRF, MSS, SZ, PM, EMY, CM, DO, MW, LEM, RB, MB, TH, SN, J-HK, MO, SWP, DKW, ET, KK, SVF, and PF contributed to data collection. JS and FV conducted statistical analyses. DMJ, TA, DD, GRF, MSS, SZ, PM, EMY, CM, DO, MW, LEM, RB, MB, TH, SN, J-HK, MO, SWP, DKW, ET, KK, SVF, JOS, JS, AMQ, FV, J-PG, WOB, and PF contributed to data interpretation. DMJ, JOS, JS, AMQ, FV, J-PG, WOB, and PF contributed to the study design. All authors contributed to the writing and review of the manuscript and approved the final version of the manuscript.
DisclosuresTA is a consultant for BMS, Boehringer Ingelheim, Roche, Merck-Schering Plough, Gilead, and Janssen.
DD has received consultancy fees from AbbVie, BI, BMS, Gilead, Idenix, and Merck.
GRF has received consultancy fees from Boehringer Ingelheim, BMS, Janssen, Gilead, Novartis, GSK, Regu-lus, Idenix, and Merck.
MSS is a member of the advisory board of AbbVie, BI, BMS, Gilead, Janssen, Merck, and Vertex; has received grants from AbbVie, BI, Gilead, Janssen, Merck, Roche, and Vertex; and consulting fees from Pfizer.
SZ has received consultancy fees from Abbvie, BMS, Boehringer Ingelheim, Gilead, Idenix, Janssen, Merck, Novartis, Presidio, Roche, Santaris, and Vertex.
PM has received consulting or advisor fee from Onyx, Gilead, Merck, and Janssen; payment, including service on speakers’ bureaus, from Onyx, Gilead, Merck, Salix, Vertex, and Janssen.
EMY has received fees from BI, Gilead, Vertex, Merck, Roche, Norvartis, Janssen, and AbbVie.
CM has served on advisory boards for Abbvie, BMS, Gilead, Janssen, and Merck; has received research grants from Astellas, Gilead, Janssen, Merck, Novartis, and Roche.
MB has served on advisory boards for Boehringer Ingelheim, Merck, Bristol Myers Squibb, Vertex, Janssen, Gilead, Abbott, GSK, and Roche; and has received lecture fees from Merck, Janssen, Boehringer Ingelheim, Bristol Myers Squibb, and Gilead.
TH has received grants from AbbVie, Boehringer Ingelheim, Bristol Myers Squibb, Eiasi, Gilead Sciences, Ikaria, Janssen, Salix Pharmaceuticals Sundise, TaiGen Biotechnology, and Vertex; and has received direct payments, including service on speakers’ bureaus, from BMS, Genentech, Gilead, and Salix.
SN received fees from Chugai Pharmaceutical, MSD, Dainippon Sumitomo Pharma, Ajinomoto Pharmaceuticals, and Otsuka Pharmaceutical.
MO has received consulting fees from Boehringer Ingelheim and Gilead.
SWP has received honoraria or consulting fees from Bayer, BMS, Gilead, Roche, and Merck.
ET has been investigator for AbbVie, BMS, BI, Gilead, Janssen, Merck, Roche, Vertex; served on advisory boards for AbbVie, BI, BMS, Gilead, Janssen, Merck, Roche, Vertex; and received speaker fees from Gilead, Janssen, Merck, Roche, Vertex.
KK has received research grants from BI, BMS, Vertex, Roche, Merck, Gilead, AbbVie, Janssen; and has served on advisory boards for Vertex, Roche, Merck, AbbVie, Gilead, Janssen.
PF is a member of advisory boards or review panels of Roche, Pfizer, Novartis, Vertex, Salix, Madaus Rottap-harm, Tibotec, Boehringer Ingelheim, and Achillion; has served as a speaker for Roche, Gilead, and Salix; and has received grants or research support from Vertex and Madaus Rottapharm.
JOS, JS, and AMQ are employees of Boehringer Ingelheim Pharmaceuticals, Inc.
FV, J-PG, and WOB are employees of Boehringer Ingelheim Pharma GmbH and Co. KG.
DMJ, DO, MW, LEM, RB, DKW, J-HK, and SVF have nothing to disclose.
 at Week 4 and < 25 IU/mL (target not detected) at week 8. PR: peginterferon alfa-2a/ribavirin. QD: once daily. SV1: STARTVerso1. SV2: STARTVerso2.")
STARTVerso1 and 2 study design. ETS: early treatment success, defined as HCV RNA < 25 IU/mL (target detected or target not detected) at Week 4 and < 25 IU/mL (target not detected) at week 8. PR: peginterferon alfa-2a/ribavirin. QD: once daily. SV1: STARTVerso1. SV2: STARTVerso2.
Baseline demographics and disease characteristics by region.
| Placebo + PegIFN/RBV | FDV 120 mg q.d. + PegIFN/RBV | FDV 240 mg q.d. + PegIFN/RBV | |
|---|---|---|---|
| Asia | n = 47 | n = 99 | n = 97 |
| Race, n (%) | |||
| Asian | 47 (100) | 99 (100) | 97 (100) |
| Country | |||
| Japan | 24 (51) | 52 (53) | 50 (52) |
| South Korea | 16 (34) | 29 (29) | 30 (31) |
| Taiwan | 7 (15) | 18 (18) | 17 (18) |
| Median age, years (range) | 54 (19–69) | 53 (20–69) | 55 (28–70) |
| Age ≥ 40 years, n (%) | 43 (91) | 84 (85) | 90 (93) |
| Mean BMI, kg/m2 (SD) | 23.3 (3.7) | 23.2 (3.2) | 23.4 (3.1) |
| HCV GT-1 subtype, n (%) | |||
| 1a | 1 (2) | 2 (2) | 2 (2) |
| 1b | 46 (98) | 97 (98) | 95 (98) |
| Baseline HCV RNA ≥ 800,000 IU/mL, n (%) | 40 (85) | 82 (83) | 73 (75) |
| IL28B GT (rs12979860), n (%) | |||
| CC | 39 (83) | 78 (79) | 78 (80) |
| CT | 8 (17) | 20 (20) | 19 (20) |
| TT | 0 | 1 (1) | 0 |
| Fibrosis stage, n (%)b | |||
| < F3 | 38 (81) | 82 (83) | 84 (87) |
| ≥ F3 | 9 (19) | 16 (16) | 13 (13) |
| Europe | n = 108 | n = 207 | n = 211 |
| Race, n (%) | |||
| Caucasian | 103 (95) | 203 (98) | 205 (97) |
| Black or African American | 2 (2) | 3 (1) | 4 (2) |
| Asian | 3 (3) | 0 | 1 (<1) |
| Othera | 0 | 1 (<1) | 1 (<1) |
| Country | |||
| Austria | 4 (4) | 13 (6) | 14 (7) |
| Belgium | 3 (3) | 19 (9) | 17 (8) |
| France | 13 (12) | 29 (14) | 30 (14) |
| Germany | 19 (18) | 24 (12) | 36 (17) |
| Portugal | 9 (8) | 13 (6) | 16 (8) |
| Romania | 13 (12) | 26 (13) | 19 (9) |
| Russian Federation | 16 (15) | 25 (12) | 25 (12) |
| Spain | 5 (5) | 25 (12) | 23 (11) |
| Switzerland | 9 (8) | 6 (3) | 15 (7) |
| United Kingdom | 17 (16) | 27 (13) | 16 (8) |
| Median age, years (range) | 46 (22-66) | 48 (20-68) | 47 (20–70) |
| Age ≥ 40 years, n (%) | 73 (68) | 148 (71) | 145 (69) |
| Mean BMI, kg/m2 (SD) | 25.1 (4.2) | 25.5 (4.2) | 25.7 (4.7) |
| HCV GT-1 subtype, n (%) | |||
| 1a | 45 (42) | 86 (42) | 89 (42) |
| 1b | 62 (57) | 120 (58) | 122 (58) |
| Other GT-1 | 1 (1) | 1 (<1) | 0 |
| Baseline HCV RNA ≥ 800,000 IU/mL, n (%) | 81 (75) | 157 (76) | 146 (69) |
| IL28B GT (rs12979860), n (%) | |||
| CC | 27 (25) | 67 (32) | 63 (30) |
| CT | 63 (58) | 111 (54) | 114 (54) |
| TT | 18 (17) | 28 (14) | 34 (16) |
| Fibrosis stage, n (%)b | |||
| < F3 | 87 (81) | 166 (80) | 171 (81) |
| ≥ F3 | 21 (19) | 40 (19) | 40 (19) |
| North America | n = 109 | n = 215 | n = 216 |
| Race, n (%) | |||
| Caucasian | 85 (78) | 171 (80) | 167 (77) |
| Black or African American | 17 (16) | 33 (15) | 35 (16) |
| Asian | 3 (3) | 5 (2) | 4 (2) |
| Othera | 4 (4) | 6 (3) | 10 (5) |
| Country | |||
| Canada | 33 (30) | 59 (27) | 55 (25) |
| USA | 76 (70) | 156 (73) | 161 (75) |
| Median age, years (range) | 51 (25–66) | 52 (18–68) | 52 (19–68) |
| Age ≥ 40 years, n (%) | 99 (91) | 190 (88) | 189 (88) |
| Mean BMI, kg/m2 (SD) | 28.4 (4.9) | 28.3 (5.4) | 28.1 (5.0) |
| HCV GT-1 subtype, n (%) | |||
| 1a | 79 (72) | 159 (74) | 162 (75) |
| 1b | 30 (28) | 55 (26) | 53 (25) |
| Other GT-1 | 0 | 1 (<1) | 1 (<1) |
| Baseline HCV RNA ≥ 800,000 IU/mL, n (%) | 88 (81) | 179 (83) | 178 (82) |
| IL28B GT (rs12979860), n (%) | |||
| CC | 28 (26) | 68 (32) | 72 (33) |
| CT | 55 (50) | 108 (50) | 102 (47) |
| TT | 25 (23) | 39 (18) | 37 (17) |
| Fibrosis stage, n (%)b | |||
| < F3 | 80 (73) | 160 (74) | 165 (76) |
| ≥ F3 | 27 (25) | 53 (25) | 44 (20) |
FibroScan® results were used to determine stage of fibrosis for patients without a liver biopsy (< F3 = < 9.5 kPa, ≥ F3 = ≥ 9.5 kPa). If a patient was indicated to have cirrhosis but had neither biopsy nor FibroScan data they were classified as having ≥ F3 fibrosis. BMI: body mass index. FDV: faldaprevir. GT: genotype. HCV: hepatitis C virus. PegIFN/RBV: peginterferon alfa-2a/ ribavirin; q.d.: once daily. SD: standard deviation.
SVR12 by region and country according to treatment group (full analysis set).
| n/N (%) | Placebo + PegIFN/RBV (n = 264) | FDV 120 mg q.d. + PegIFN/RBV (n = 521) | FDV 240 mg q.d. + PegIFN/RBV (n = 524) |
|---|---|---|---|
| North America | 49/109 (45) | 135/215 (63) | 129/216 (60) |
| Canada | 17/33 (52) | 40/59 (68) | 41/55 (75) |
| USA | 32/76 (42) | 95/156 (61) | 88/161 (55) |
| Europe | 53/108 (49) | 160/207 (77) | 164/211 (78) |
| Asia | 29/47 (62) | 87/99 (88) | 85/97 (88) |
FDV: faldaprevir. PegIFN/RBV: peginterferon alfa-2a/ribavirin. q.d.: once daily. SVR12: sustained virologic response (HCV RNA < 25 IU/mL target not detected) 12 weeks after completion of therapy.
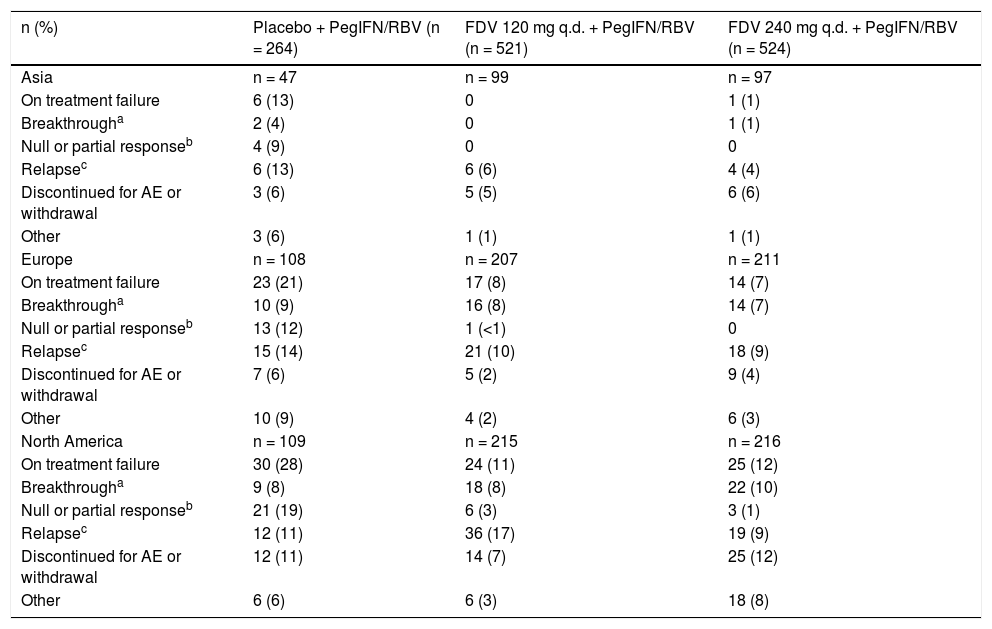
Treatment failure by region.
| n (%) | Placebo + PegIFN/RBV (n = 264) | FDV 120 mg q.d. + PegIFN/RBV (n = 521) | FDV 240 mg q.d. + PegIFN/RBV (n = 524) |
|---|---|---|---|
| Asia | n = 47 | n = 99 | n = 97 |
| On treatment failure | 6 (13) | 0 | 1 (1) |
| Breakthrougha | 2 (4) | 0 | 1 (1) |
| Null or partial responseb | 4 (9) | 0 | 0 |
| Relapsec | 6 (13) | 6 (6) | 4 (4) |
| Discontinued for AE or withdrawal | 3 (6) | 5 (5) | 6 (6) |
| Other | 3 (6) | 1 (1) | 1 (1) |
| Europe | n = 108 | n = 207 | n = 211 |
| On treatment failure | 23 (21) | 17 (8) | 14 (7) |
| Breakthrougha | 10 (9) | 16 (8) | 14 (7) |
| Null or partial responseb | 13 (12) | 1 (<1) | 0 |
| Relapsec | 15 (14) | 21 (10) | 18 (9) |
| Discontinued for AE or withdrawal | 7 (6) | 5 (2) | 9 (4) |
| Other | 10 (9) | 4 (2) | 6 (3) |
| North America | n = 109 | n = 215 | n = 216 |
| On treatment failure | 30 (28) | 24 (11) | 25 (12) |
| Breakthrougha | 9 (8) | 18 (8) | 22 (10) |
| Null or partial responseb | 21 (19) | 6 (3) | 3 (1) |
| Relapsec | 12 (11) | 36 (17) | 19 (9) |
| Discontinued for AE or withdrawal | 12 (11) | 14 (7) | 25 (12) |
| Other | 6 (6) | 6 (3) | 18 (8) |



