






Failure of cholesterol homeostasis in the body can lead to cholesterol gallstone disease, the most common and costly gastrointestinal disease. The primum movens in cholesterol gallstone formation is the hypersecretion of hepatic cholesterol; this condition leads to bile chronically surpersaturated with cholesterol which is prone to rapid precipitation as cholesterol crystals in the gallbladder. Essential topics reviewed here deal with pathways of biliary lipid secretion, cholesterol solubilization and crystallization in bile, according to recent advances. Main in vivo events in cholesterol gallstone disease are also described.
Abbreviations: AQPs, aquaporins; BSEP, bile salt export pump; DPPC, dipalmitoyl phosphatidylcholine; CSI, cholesterol saturation index; EYPC, egg-yolk phosphatidylcholine; MDR, multi-drugresistant; MMC, migrating motor complex; PFIC, progressive familial intrahepatic cholestasis; PL, phospholipids; SM, sphingomyelin
IntroductionFailure of cholesterol homeostasis in the body can result in cholesterol gallstone disease, the most common and costly gastrointestinal disease.1,2 The estimated prevalence of cholelithiasis in adult population is 10-15%.3 Although there are three predominant types of stones: cholesterol, brown pigment and black pigment, in countries using western-style diet, gallstones are made of 75-80% cholesterol3 in up to 80% of patients with gallstones.4
The classical paradigma on the pathogenesis of cholesterol gallstones relies on the coexistence of at least 3 defects: hypersecretion of hepatic cholesterol resulting in chronically surpersaturated bile, increased crystallization of biliary cholesterol and gallbladder stasis.6-9 The study of physical-chemical factors and pathways leading to cholesterol crystallization in bile has clinical relevance. A better understanding of early events in gallstone formation, in fact, plays a key role for the identification of potential factors delaying or preventing precipitation of cholesterol crystals and gallstone formation in bile.
In this review, current opinions on the pathogenesis of cholesterol gallstones with respect to lipid secretion, cholesterol solubilization and crystallization in bile will be discussed.
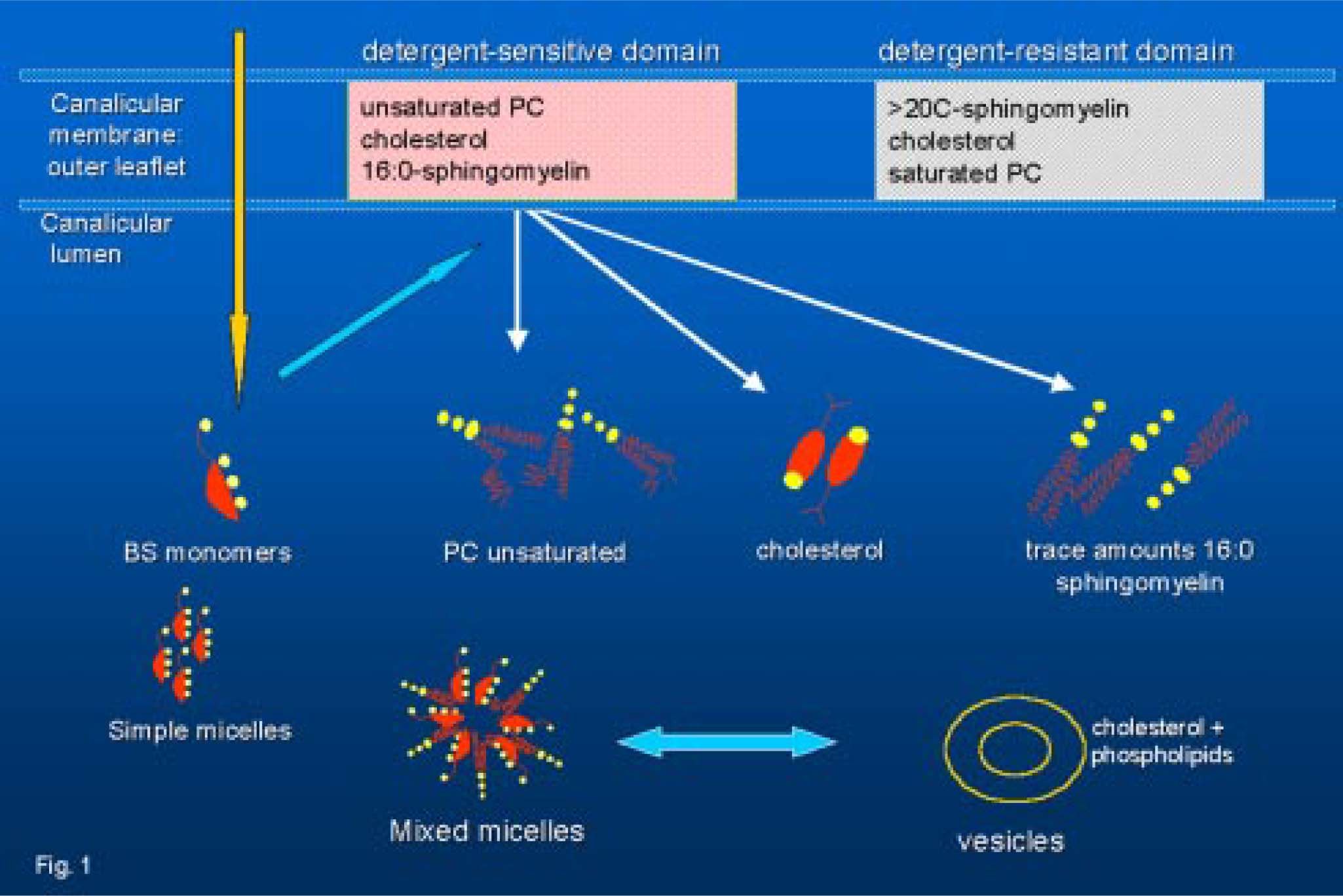
Secretion of lipids in bileThe hepatocyte plasma membrane is functionally divided into a canalicular (or apical) region adjacent to the lumen of the bile canaliculus and a sinusoidal (or basolateral) region in close contact with sinusoidal blood. The process of bile secretion initiates at the hepatic level in the canaliculus, which is a specialized luminal space formed by two hepatocytes. Although the canalicular region comprises only 10-15% of the total plasma membrane, it plays a crucial role in the process of nascent bile formation and biliary secretion of bile salts, water, phospholipids and cholesterol. Bile is a complex fluid in which over 95% is water10,11 mixed with three classes of lipids (i.e. bile salts, cholesterol, phospholipids), proteins, bilirubin, and organic anions.5 Bile formation starts with active secretion of solutes, followed by osmotic attraction of water. The hepatocyte is the major site for cholesterol synthesis and its elimination in bile. Hepatic secretion of bile salts and cholesterol into bile is the basis for the elimination of excess cholesterol from the body. Molecular mechanisms of the vectorial transport of biliary constituents into the bile are now better understood12,13(Figure 1). Canalicular secretion of bile is a primary active transport mediated by a series of ATP-binding cassette (ABC) transporters.14 They comprise the sister of P-glycoprotein (Spgp)15,16 also known as the bile salt export pump (BSEP, the major canalicular bile salt export pump of mammalian liver), the canalicular multidrug resistance protein MRP2 (cMOAT/ cMRP) for transport of anionic conjugates such as glucuronide and glutathione conjugates17,18 the canalicular multidrug resistance P-glycoprotein MDR1 for excretion of hydrophobic drugs, amphiphilic organic cations, toxins, metabolites, steroid hormones, hydrophobic peptides or glycolipids.19ABC transporters also include the human MDR3 P-glycoprotein20 (corresponding to the murine mdr2 P-glycoprotein21) which functions as a “flippase” of phosphatidylcholine molecules from the inner to the outer leaflet of the canalicular membrane.22 Lipid transporters in bile and relative genes are listed in Table I. The relationship between bile salt and lipid secretion is curvilinear; the secretion of both phospholipids and cholesterol plateaus at high bile salt secretion rates.23 However, phospholipid secretion rate is always higher than that of cholesterol. Amount of the bile salts-induced biliary lipid secretion is positively related to the hydrophobicity of secreted bile salt species.24 Secretion of “flipped” phosphatidylcholine from the outer leaflet into the canalicular lumen means mainly formation of vesicles from the external hemileaflet of the canalicular membrane.25 Following their secretion into the canalicular lumen, detergent bile salts directly micellize considerable amounts of lipid from the membrane.26,27 Experiments with mdr2 deficient mice (i.e. phosphatydilcholine not secreted) show that cholesterol secretion can occur without phospholipid secretion; in this model the infusion of the hydrophobic bile salt deoxycholate is sufficient for the extraction of cholesterol from the canalicular membrane.26
, and detergent-resistant laterally separated domains (sphingomyelin with long ≥ 20 C atoms saturated acyl chains, disaturated phosphatidylcholine species and cholesterol (modified according to81)). PC, phosphatidylcholine.")
Current views on lipid secretion in bile.81 The outer leaflet of the hepatocyte canalicular membrane may contain detergent-sensitive domains (with bile-destined phosphatidylcholine species, that have unsaturated acyl chains at the sn-2 position, plus small amounts of 16:0 sphingomyelin and cholesterol), and detergent-resistant laterally separated domains (sphingomyelin with long ≥ 20 C atoms saturated acyl chains, disaturated phosphatidylcholine species and cholesterol (modified according to81)). PC, phosphatidylcholine.
Lipid transporters involved in bile formations and the phenotype of their mutations.
| Name | Gene | Function | Mutation |
|---|---|---|---|
| MDR-3 | ABCB4 | PC flippase from inner to outer leaflet of the hepatocyte canalicular membrane | PFIC-3 |
| BSEP | ABCB11 | Bile salt export pump across the hepatocyte canalicular membrane | PFIC-2 |
| ABCG5 | ABCG5 | Phytosterols, cholesterol? | Sitosterolemia |
| ABCG8 | ABCG8 | Cholesterol? | unknown |
Adapted from Oude Elferink and Groen28
Legend: BSEP, bile salt export pump; MDR, multi-drug-resistant; PC, phosphatidylcholine; PFIC, progressive familiar intrahepatic cholestasis
These findings points to an alternative path of cholesterol secretion. Researches are focusing on the so-called “cholesterol pumping system”.28 One theory has considered the possibility that the ABC transporter ABCA1 could play a direct role in cholesterol secretion in bile. ABCA1 regulates serum HDL cholesterol levels and is considered to control the first step of reverse cholesterol transport from the periphery to the liver.29-31 Recently, Groen et al.32 have shown that Abca1-/- knockout mice have preserved biliary secretion rates of cholesterol, bile salts, and phospholipid as compared with wild-type animals, in spite of absence of HDL. It appears that plasma HDL levels and ABCA1 activity do not control net cholesterol transport from the periphery via the liver into bile, indicating that ABCA1 has no direct role in cholesterol secretion in bile.
Recently, a possible role for other two ABC trasporters, namely ABCG5 and ABCG8 (both expressed in the intestine and in the liver) in pumping cholesterol molecules in bile, has been advocated.28,33 A direct evidence suggests that the two transporters might be involved in the elimination of plant sterols. Accumulation of plant sterols, in fact, has been observed in patients with sitosterolemia, a recessively inherited rare disease in which the disease locus is localized to chromosome 2p21 and ABCG5 and ABCG8 genes are involved.33,34
Cholesterol carriers are necessary in bile, as cholesterol is poorly soluble in an aqueous environment (Figure 2). Phenomena of lipid aggregation in bile start immediately after secretion; cholesterol is solubilized in bile in mixed micelles by bile salts and phospholipids. Phosphatidylcholine (PC) is the major phospholipid in bile (> 95% of total: mainly 16:0 acyl chains at the sn-1 position and mainly unsaturated (18:2 > 18:1 > 20:4) acyl chains at the sn-2 position35). In the canalicular space also cholesterol/ PC vesicles exist (ratio ~0.3). During concentration of bile in the bile ducts, however, the cholesterol/PC vesicular ratio increases (> 1), due to progressive micellization of PC by bile salts. In the gallbladder, further concentration of bile results in vesicles aggregates and fusion into multilamellar supersaturated cholesterol/PC vesicles (ratio > 1); such vesicles are unstable due to cholesterol-rich microdomains, prone to precipitate as cholesterol crystals.36,37 Events are greatly accelerated in the case of cholesterol supersaturation (see below). The possibility that cholesterol crystals precipitate also from “supersaturated” micelles, has been advocated.38
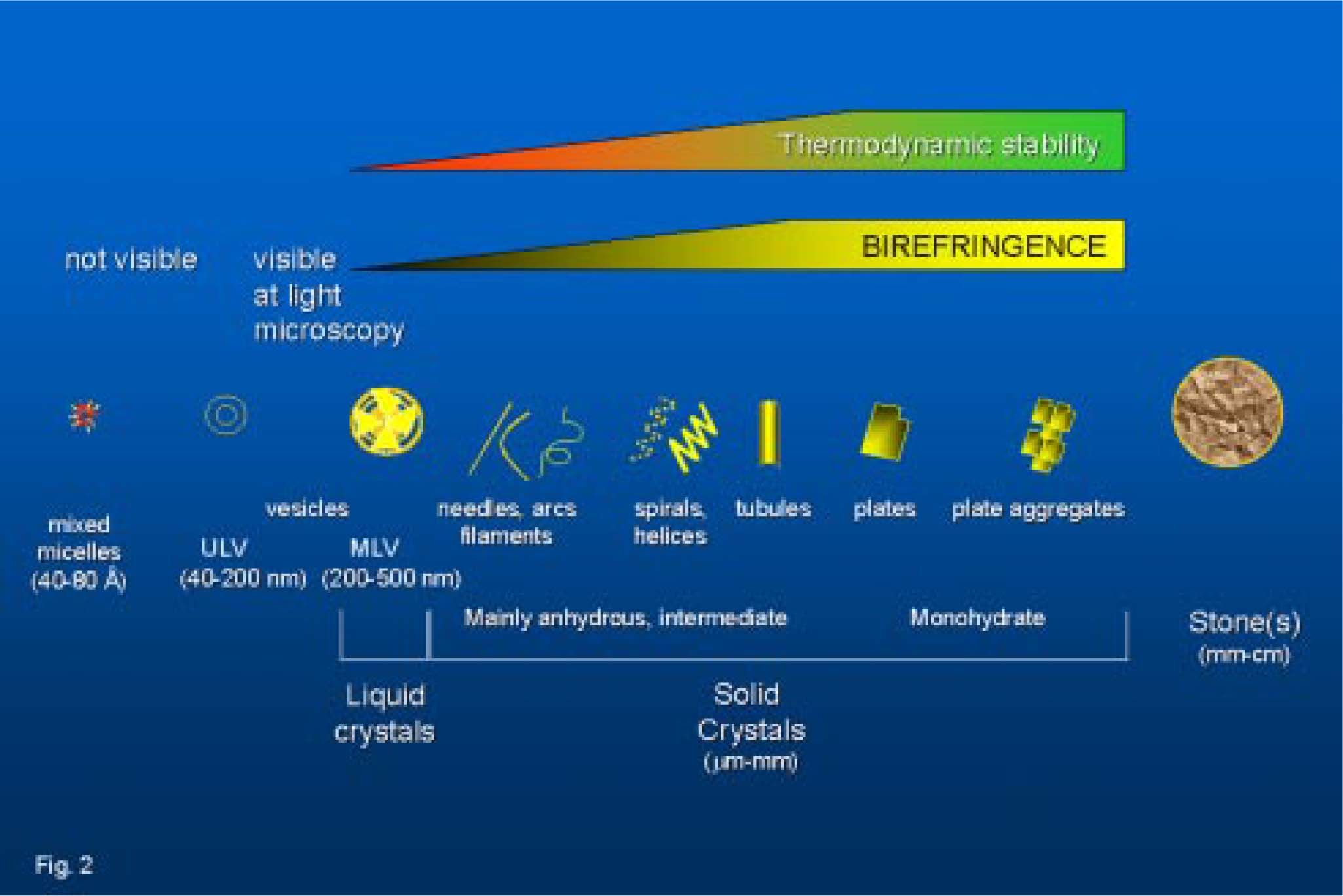
From cholesterol solubilization to cholesterol crystallization.")
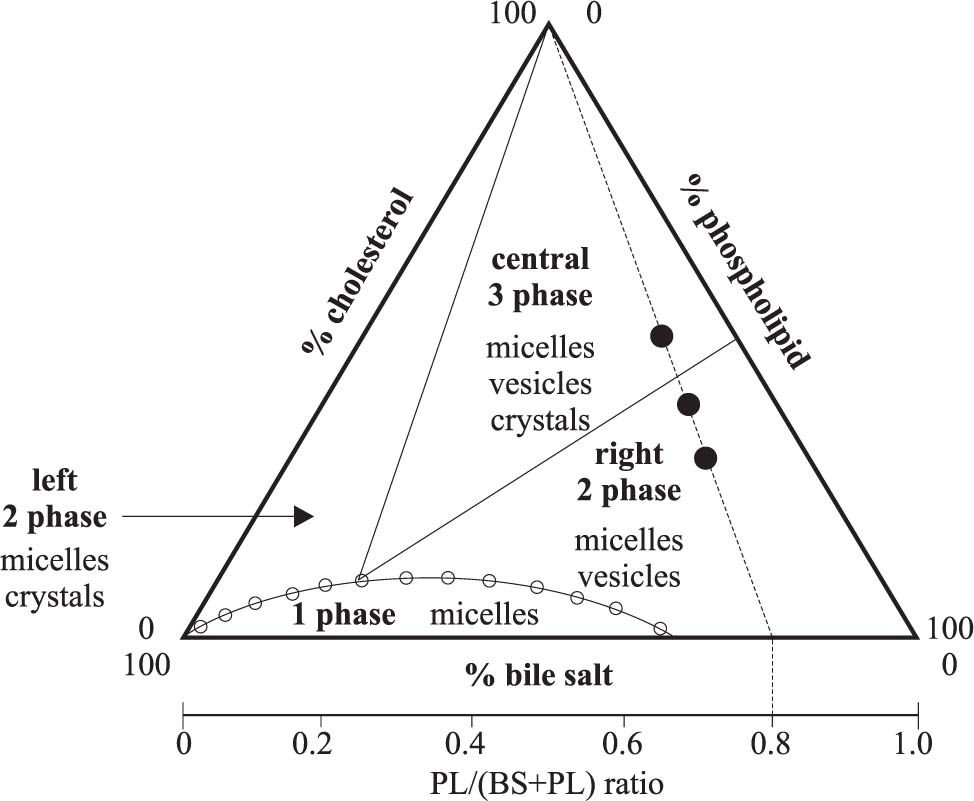
The conditions when cholesterol crystallization occurs are depicted within the ternary phase diagram in which the behaviour of cholesterol-bile salt-phospholipid aggregates in aqueous solutions are shown (Figure 3). Appearance and transition of micelles, vesicles, and cholesterol crystals are studied between metastable supersaturated systems and thermodynamic equilibrium (at fixed relative total lipid concentration, temperature, and pH). Wang & Carey39 demonstrated that the ternary phase diagram contains a one-phase zone (only micelles), a left two-phase (micelles and cholesterol crystals-containing) zone, a central three-phase (micelles, vesicles and cholesterol crystals-containing) zone and a right two-phase (micelles and vesicles-containing) zone. Accurate quantification of such phases requires isolation of micelles and vesicles by gel filtration40-43 and repeated observation of crystal growth.39 Distinct sequences of cholesterol crystallization occur in the left two-phase and the central three-phase zones, involving anhydrous cholesterol crystals (needles, arcs, tubules, spirals, mass density ~1.03 g/dL) firtsly described in model biles by Konikoff et al. in 199244 and mature rhomboid birefringent plate-like cholesterol monohydrate crystals (mass density ~1.45 g/dL). Table II depicts the characteristics of different cholesterol crystal forms observed in bile. We also studied this topic45 and recently we compared four independent methods to assess cholesterol crystallization in model biles i.e.light microscopy, nephelometry, spectrophotometry and biochemical assay; we found that morphology and size of cholesterol crystals strongly affect turbidimetric estimates of crystal mass. In principle, chemical measurement of crystal mass should be the method of choice for exact quantitation of cholesterol crystallization within the phase diagram.46
 and defines the limits of cholesterol solubility. Phase boundaries depend on relative composition of lipids, total lipid concentration (7.3 g/dL), and temperature (37°C). Each triangular apex depicts biles made of a single lipid. Points plotting within the diagram depict biles with different composition. Depicted are a onephase (micellar) zone at the bottom, a left two-phase zone (containing micelles and crystals), a central three-phase zone (containing micelles, vesicles and crystals) and a right two-phase zone (containing micelles and vesicles). The cholesterol saturation index (CSI) has its maximum normal value set at 1 (open dots at the micellar-phase boundary) and is defined as the ratio between molar percentage of cholesterol and the maximum micellar solubility of cholesterol. It can be seen from the diagram that for CSI > 1, biles must contain at least one additional phase of cholesterol at equilibrium. On the basal axis is also depicted an axis with PL/(BS+PL) ratios. The three theoretical model biles (•) plotting on interrupted line have identical PL/(BS+PL) ratios (in this case PL/ (BS+PL) ratio = 0.8).")
The ternary equilibrium phase diagram39 shows relative concentrations of the three major biliary lipids (cholesterol, phospatidylcholine -PL, bile salts -BS,) and defines the limits of cholesterol solubility. Phase boundaries depend on relative composition of lipids, total lipid concentration (7.3 g/dL), and temperature (37°C). Each triangular apex depicts biles made of a single lipid. Points plotting within the diagram depict biles with different composition. Depicted are a onephase (micellar) zone at the bottom, a left two-phase zone (containing micelles and crystals), a central three-phase zone (containing micelles, vesicles and crystals) and a right two-phase zone (containing micelles and vesicles). The cholesterol saturation index (CSI) has its maximum normal value set at 1 (open dots at the micellar-phase boundary) and is defined as the ratio between molar percentage of cholesterol and the maximum micellar solubility of cholesterol. It can be seen from the diagram that for CSI > 1, biles must contain at least one additional phase of cholesterol at equilibrium. On the basal axis is also depicted an axis with PL/(BS+PL) ratios. The three theoretical model biles (•) plotting on interrupted line have identical PL/(BS+PL) ratios (in this case PL/ (BS+PL) ratio = 0.8).
Crystal forms of cholesterol in bile
| Form | Crystal configuration | Crystal habit1 | Unit cell | Mass density g/dL | Birefringence (polarizing light) | Thermodynamic stability at 37°C2 |
|---|---|---|---|---|---|---|
| Monohydrate | Triclinic | Romboid, plate-like, fixed angles (79.2 and 100.8 degrees), ften notched corner. Better observed at continuous high humidity | 8 molecules cholesterol 8 molecules water | 1.045 | +++ | stable |
| Anhydrous | Triclinic | Needle-like | 8 molecules cholesterol (low (< 31.6°C) temperature) 16 molecules cholesterol (high (> 31.6°C) temperature) | 1.03 | —+ | unstable |
| Intermediate(mainly anhydrous) | Filaments, arcs, needles, helices, tubules | ~1.03 | + | metastable |
Note:1 depending on hydration status and speed of crystallization; 2indicates that crystal structure is less favourable thermodynamically82
The study of changes within the ternary phase diagram has pathophysiogical relevance for the prevention of cholesterol crystallization and, ultimately, of cholesterol gallstone formation. When bile is diluted (i.e. lower total lipid concentration) or enriched with disaturated phospholipids or hydrophilic bile salts, the micellar zone is smaller and the right two-phase (i.e. vesicles and micelles-containing) zone expands to the left at the expense of the crystals-containing (i.e. central three-phase and left two-phase) zones. The opposite is true with concentrated bile or biles enriched with unsaturated phospholipids or hydrophobic bile salts. We found that in excess of bile salts (i.e. low phospholipid/(bile salts/phospholipid) ratios-referred to as PL/(BS+PL) ratios ~ < 0.2), crystals precipitate at fast rates forming intermediate anhydrous cholesterol crystals and mature rhomboid plate-like cholesterol monohydrate crystals.46 At higher amounts of phospholipids, large amounts of cholesterol are solubilized in vesicles together with phospholipids and crystal precipitation proceeds at slower rates as mainly mature cholesterol monohydrate. In case of excess phospholipids (high PL/ (BS+PL) ratios), solid cholesterol crystals do not occur, and cholesterol is mainly solubilized in vesicular phases. We also showed that phospholipids can interfere within model systems plotting in the left two-phase or central three-phase (crystal-containing) zones. Indeed, speed and extent of crystallization increases with dipalmitoyl phosphatidylcholine (DPPC) or sphingomyelin (SM), as compared with egg-yolk phosphatidylcholine (EYPC).45 Disaturated phosphatydilcholine (PC) species inhibit crystallization, and PC species with unsaturated acyl chains at the sn-2 position progressively promote crystallization at increasing unsaturation.47-49 Thus, compared to EYPC-containing systems,39 in SM-or DPPC-containing systems50 the right two-phase (vesicles and micelles-containing) zone is greatly expanded to the left at the expense of the crystals-containing (central three-phase and left twophase) zones.
It has been suggested that dietary modification towards more saturated biliary phospholipids might prevent gallstone formation in humans, but effects of dietary modification are expected to be relatively small. In a clinical study, biliary cholesterol crystallization or lipid solubilization did not change with dietary modification.51
In our studies, the hydrophilic bile salt tauroursodeoxycholate prevented cholesterol crystallization and protected against more hydrophobic bile salt-induced crystallization;45,52 this is consistent with previous data.53 Apparently, solubilization of cholesterol in vesicular phases (obtained by enlargement of the central three-phase zone) is a prerequisite for reduced crystallization by tauroursodeoxycholate. Ursodeoxycholate or its tauro-conjugate are used as oral litholitic agents in cholesterol gallstones. When ursodeoxycholate was fed to gallstone patients (~12 mg/Kg b.w./d during 1 mo.), we found that ex vivo incubated fresh bile contained cholesterol crystals which decreased in size or even disappeared at prolonged observation, while ultrafiltered (isotropic) incubated bile remained totally crystal-free.54 Newly synthesized fatty acid bile acid conjugates (FABAC) have been shown to dissolve pre-existing cholesterol crystals55 and to dissolve stones when given orally to gallstone-susceptible C57J/L mice56 but more studies are necessary to translate such observations into clinical practice.
Cholesterol gallstone disease: an overview of events in vivoCholesterol gallstone disease is a multifactorial condition, and in vivo the events are even more complex. Liver (secretion), gallbladder (concentration) and intestine (absorption) are the three organs handling biliary lipids which play a key role in the pathogenesis of the cholesterol gallstone disease. Table III depicts the most important factors which deserve attention. In the liver increased cholesterol synthesis due to increased HMGCoA activity has been reported.57 In bile, cholesterol precipitation in crystal forms6,44,58 as well as increased bile salt hydrophobicity59 are present, together with increased secretion of pronucleating proteins,54 and especially mucin.60 The combination of factors such as cholesterol supersaturation, bile salt hydrophobicity, and pronucleating proteins has been recently investigated by van Erpecum et al.61 in mice susceptible to cholesterol gallstone formation (i.e. C57L inbred mice). It has been demonstrated that both cholesterol supersaturation and crystallization plus increased bile salt hydrophobicity and mucin concentration are early pathogenic events; in vitrocrystallization-promoting immunoglobulins and aminopeptidase-N do not appear to be major factors in murine gallstone pathogenesis. Motility defects of gallbladder, both in vivo62,66 and ex vivo,62,67,68 leading to gallbladder stasis are also important risk factors for gallstone disease. Finally, intestine may play an important role,69 since intestinal motiltiy disorders are common in gallstone patients,70 with an incoordination of the migrating motor complex (MMC), the housekeeper of the in-testine.71 In figure 4 are reported some of the current concepts on the pathogenesis of cholesterol gallstones.
Major in vivo events in cholesterol gallstone disease.
| Events | Mechanisms | Ref. |
|---|---|---|
| Increased cholesterol synthesis | Increased HMGCoA activity | 57 |
| Cholesterol crystallization | Precipitation of cholesterol in supersaturated bile | 6,44,58 |
| Increased bile salt hydrophobicity | Increased deoxycholate production | 59 |
| Protein secretion from gallbladder wall Gallbladder stasis | Increased pronucleating mucin concentration | 60 |
| Gallbladder stasis | Absence of gallbladder volume fluctuations in fasting state; | |
| Dissociation from MMC; | 70 | |
| Impaired gallbladder postprandial emptying; | ||
| Reduced postprandial turnover of bile | 63-65 | |
| Defective contractility of gallbladder strips | Smooth muscle disfunction due to miotoxic cholesterol accumulation | 62-67 |
| Slow intestinal transit | Impairment of MMC | 71 |
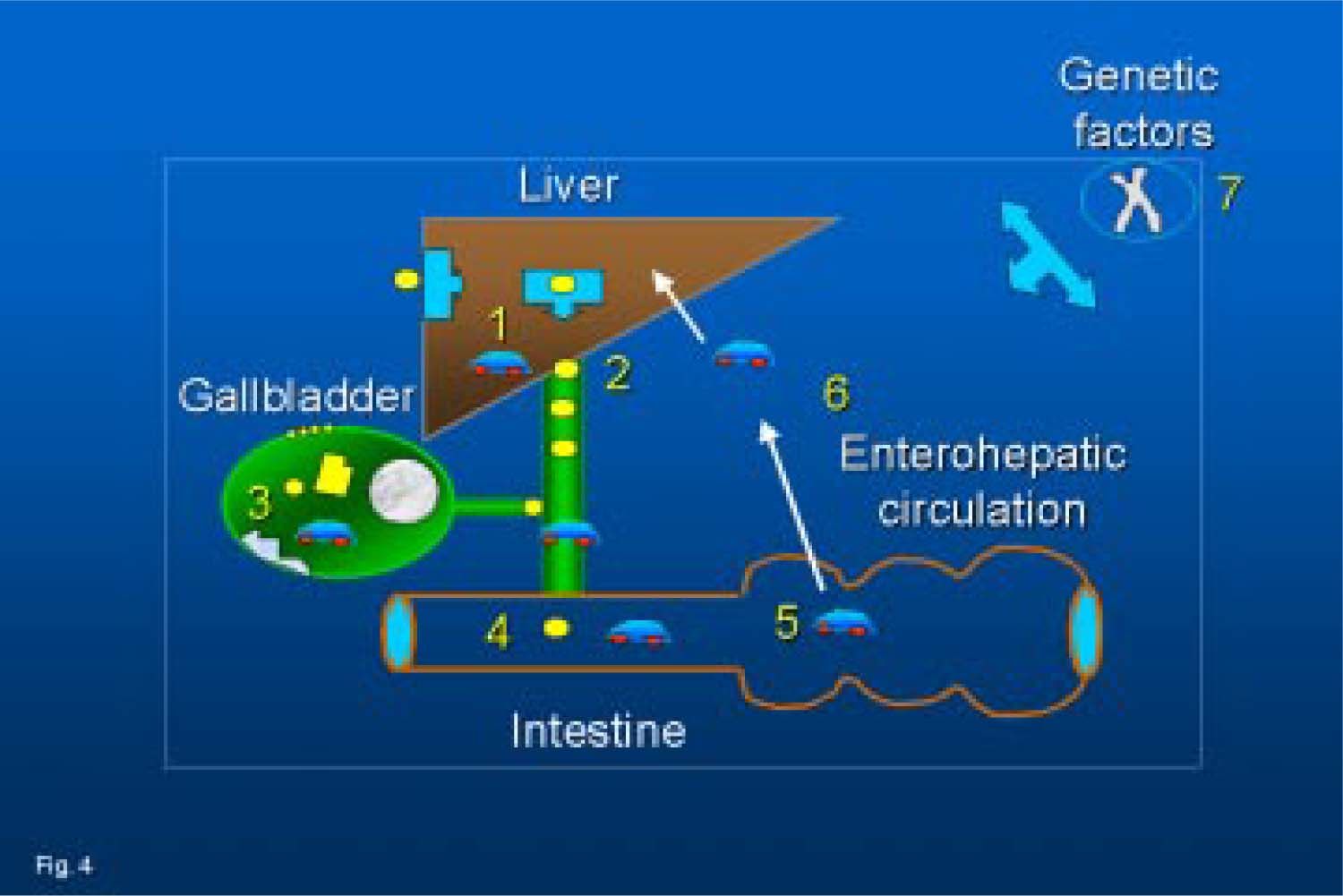
; 4, intestinal hypomotility (and delayed transit of bile); 5, increased bacterial production of deoxycholate; 6, increased liver uptake and biliary (re)secretion of deoxycholate; 7, the genetic background can influence several of the above mentioned mechanisms.")
Current concepts on the pathogenesis of cholesterol gallstones: 1, cholesterol hypersecretion by the liver; 2, sustained hypercholesterobilia; 3, events in the gallbladder (colesterol incorporation into the sarcolemma leading to defective smooth muscle contractility, increased mucin secretion, increased deoxycholate, cholesterol crystallization, stone growth); 4, intestinal hypomotility (and delayed transit of bile); 5, increased bacterial production of deoxycholate; 6, increased liver uptake and biliary (re)secretion of deoxycholate; 7, the genetic background can influence several of the above mentioned mechanisms.
In conclusion, current concepts on pathogenic factors in cholesterol gallstones are schematically depicted in figure 4. Excess biliary cholesterol represents the primum movens and this condition anticipates increased incorporation of cholesterol into the sarcolemma (hypomotile gallbladder and delayed intestinal transit), sluggish enterohepatic recir-culation of bile salts, increased hepatic reuptake and biliary secretion of the hydrophobic bile salt deoxycholate. As a result, more cholesterol secreted in bile becomes available for precipitation as crystals. Events leading to cholesterol crystallization need to be studied both in vitro by artificial model biles plotting in the ternary equilibrium phase diagram and in human bile ex vivo. Cholesterol gallstone formation may be partly genetically determined, as suggested by both epidemiologic studies,72 family surveys,73 and clinical stud-ies.74-78,79 The role of genes in the pathogenesis of gallstones has been recently reviewed.80 In the murine model, candidate genes for cholesterol gallstones have been identified: defects are found for several intracellular biliary proteins (hepatocyte cytosol); membrane lipid transport proteins (hepatocytes and cholangiocytes), transcription factors (membrane-bound and ligand-activated), lipoprotein receptors and intracellular lipid transporters. Changes in genes for the conformation/activity of the gallbladder smooth muscle CCK receptor and changes in the expression of gallbladder epithelial mucin might represent further pathogenic factors for cholesterol gallstone formation. More research, however, is needed to unravel remaining mechanisms of cholesterol gallstone disease.



