





Blood glucose fluctuates severely in the diabetes (DM) and tumor microenvironment. Our previous works have found Hepatitis B virus X protein (HBx) differentially regulated metastasis and apoptosis of hepatoma cells depending on glucose concentration. We here aimed to explore whether HBx played dual roles in the angiogenesis of hepatocellular carcinoma varying on different glucose levels.
Materials and MethodsWe collected conditioned medium from HBx-overexpressing cells cultured with two solubilities of glucose, and then applied to EA.hy926 cells. Alternatively, a co-culture cell system was established with hepatoma cells and EA.hy926 cells. We analyzed the angiogenesis of EA.hy926 cells with CCK8, wound-healing, transwell-migartion and tube formation experiment. ELISA was conducted to detect the secretion levels of angiogenesis-related factors. siRNAs were used to detect the P53-VEGF axis.
ResultsHBx expressed in hepatoma cells suppressed VEGF secretion, and subsequently inhibited the proliferation, migration and tube formation of EA.hy926 cells in a high glucose condition, while attenuating these in the lower glucose condition. Furthermore, the p53-VEGF axis was required for the dual role of HBx in angiogenesis. Additionally, HBx mainly regulated the nuclear p53.
ConclusionsThese data suggest that the dual roles of HBx confer hepatoma cells to remain in a glucose-rich environment and escape from the glucose-low milieu through tumor vessels, promoting liver tumor progression overall. We exclusively revealed the dual role of HBx on the angiogenesis of liver tumors, which may shed new light on the mechanism and management strategy of HBV- and DM-related hepatocellular carcinoma.
Liver cancer is the 6th most common cancer and one of the most fatal cancers [1]. Among them, Hepatocellular Carcinoma (HCC) accounts for 90 % of cases [1]. HCC is a hyper-vascular solid tumor in which tumor-induced angiogenesis plays a vital role in tumor proliferation and spread [2]. To trigger the angiogenic switch, a set of angiogenic factors were secreted from hepatoma cells and bound to specific receptors on the cellular membrane of endothelial cells [3,4]. The activated endothelial cells subsequently process to proliferate, migrate and ultimately assemble into new tumor capillary blood vessels [3,4]. The development of neovascularization is initiated by stresses, like hypoxia [5], and requires coordinated genetic modulation like inhibition of p53 expression to shift the balance between pro- and anti-angiogenic factors of angiogenesis [6,7]. During the angiogenesis process, vascular endothelial growth factor (VEGF) acts as the major stimulator in HCC, which is also the most widely studied factor so far [3]. Tumor cells-derived VEGF binds to two cell surface receptor tyrosine kinases on the endothelial cells’ surface, VEGFR, and then triggers a phosphorylation cascade, resulting in elevated mitosis in endothelial cells and increased vascular permeability [3].
VEGFs are shown to be regulated by many viruses including hepatitis B virus (HBV) infection [8], one of the major causative factors for HCC. The viral genome encodes the Hepatitis B virus X protein (HBx), which is a vital mediator of the initiation and development of HBV-related HCC by transcriptional transactivation and inducing a repertoire of signaling pathways [9]. HBx alone could elicit an angiogenic response [10]. HBx stimulates the VEGF promoter resulting in increased VEGF mRNA expression in hepatoma cells [10,11]. In clinical investigations, HBx-positive HCCs compared to non-HBV-related HCC tissues also exhibit higher VEGF expression levels and mean micro-vessel density [8]. HCC angiogenic guard involves various factors in building up a network. The mechanisms underlying the upregulation of VEGF and angiogenesis in HCC by HBx are far from being well understood.
Besides the well-known diversity of the HBx effects on HCC development, cumulative studies have found that the roles of HBx are usually contrary partially due to the context-dependent feature [12]. HBx plays a pro-apoptotic or anti-apoptotic effect on primary hepatocytes depending on the activation of transcription factor NFκB [12]. Studies in HBx-transgenic mice show that HBx simultaneously promotes the expression of pro-apoptotic BAX and anti-apoptotic Bcl-xL [12]. It appears that HBx needs both of the contrary functions to elegantly cope with various hepatocyte environments. Rare studies have explored the effect of glucose on the Hepatitis B virus X protein function. Our previous study has shown that HBx prevents hepatoma cells in starvation from apoptosis by elevating mitophagy while promoting apoptosis when cells are in an adequate medium [13]. In addition, we also found that HBx differentially regulated metastasis of hepatoma cells according to different glucose concentrations (to be published). We wondered whether HBx played dual roles in angiogenesis in response to different glucose levels.
The liver, as the center of glucose synthesis and catabolism in the body, creates a glucose concentration gradient along the Porto-central axis [14]. Tumors are characterized by nutrient avid [15]. Due to the differences in nutrient supply by vascular, liver tumor interstitium are uneven in glucose distribution. The normal blood sugar concentration is about 5.5mM. In many cases, hyperglycemia accelerates the progression of a tumor by upregulating the proliferation and migration of tumor cells [16]. Evidence from hepatoma cell models has proved that high glucose (25-50mM) induced c‑Met activation promotes aggressive phenotype [17]. Additionally, diabetes (DM) characterized by dysregulation in the blood glucose level, confers complex nutrient microenvironment to tumor development. There are limited investigations about the biological behavior of hepatoma cells under different glucose concentrations so far, which may provide new insights into the mechanisms of HCC development.
In this study, we focused on the effects and mechanisms of HBx on the interaction between hepatoma cells and endothelial cells, especially comparing those under the low glucose environment with the high glucose condition, aiming to identify the dual role of HBx on angiogenesis.
2Material and methods2.1Cell culture and plasmidsHepG2 cells, obtained from the American Type Culture Collection (ATCC, USA), Huh7 cells and SMMC-7721 cells, provided by Cell Bank (Chinese Academy of Sciences), Hep3B cells and EA.hy926 cells, obtained from Cell Bank (Guangzhou Cellcook Biotech Co., Ltd, China), were respectively maintained in low-DMEM medium (Hyclone, USA) supplemented with 12 % fetal bovine serum (Hyclone, USA), adding extra 100 × MEM Non-Essential Amino Acids Solution (NEAA, gibco, USA) and GlutaMAX (gibco, USA). For a high glucose condition, D-(+)-Glucose solution (gibco, USA) was added to cultured media, reaching 30mM of the glucose concentration. To inhibit the ubiquitin-proteasome pathway, hepatoma cell lines were treated with 10μM MG132 (Sigma, USA) for 6hr or lactacystin (Sigma, USA) for 24hr. HBx-expressing plasmids pcDNA3.1-flag-HBx (adw subtype) and pcDNA3.1-vector, as well as HBx-expressing adenovirus HBx-Ad (adw subtype) and vector-adenovirus, were all gifts from professor Xu Lin (Key Laboratory of Ministry of Education for Gastrointestinal Cancer, Fujian Medical University, China). Plasmids transfection was conducted by Lipofectamine™ 3000 (Invitrogen, USA).
2.2Quantitative realtime-PCRTotal RNA extraction and Quantitative RT-PCR were performed as described previously [13]. The primers used are shown in Table 1. The relative gene expression levels were calculated with the method of 2^(-∆∆Ct)values.
Primers used in RNA extraction.
Total protein extraction and western blot analysis were performed as previously described [13], and the primary antibodies used are as follows: rabbit anti-p53 antibody (USA, cell signaling technology), rabbit anti-HBx antibody (USA, sigma), rabbit anti-N-cad antibody (USA, CST), mouse anti-E-cad antibody (USA, CST), rabbit anti-VEGF-A antibody (USA, sigma), rabbit anti-Lamin B1 antibody (USA, CST), rabbit anti-β-tubulin (USA, CST), mouse anti-β−actin antibody (USA, sigma).
2.4Cell proliferation assay (Cell Counting Kit-8, CCK8)We used Cell Counting Kit-8 (dojindo, Japan) to determine cell viability in cell proliferation assays. We strictly followed the kit manual and performed it in a 96-well plate with 5-6 repeats in each group. The reagent mixture was incubated at 37°C for 1-3hr according to cell type.
2.5Wound healing assayCells were seeded in a 6-well plate and cultured with a conditioned medium overnight until overgrown. A scratch was created with a toothpick. The cell migration was photographed under a microscope at a certain time point after the scratch.
2.6Transwell chamber migration assayHepatoma cells were seeded in the lower compartment of the transwell chamber (Corning, USA), and EA. hy926 cells were seeded in the upper transwell inserts. After co-culturing the cells for 24 h, cells that had migrated through the monolayer and attached at the outside of the membrane were fixed with 4 % paraformaldehyde and stained with 0.1 % crystal violet for 30 min at room temperature. The migration cells were photoed and counted under a microscope.
2.7Tube formation assayA 96-well plate was precoated with growth factor-reduced matrigel (BD Biosciences, USA) and then seeded with EA.hy926 cells that had starved with serum-free medium for 12hr ahead. After culturing the cells at 37°C for 4-6hr, the tube formation was photographed under a microscope. The number of tubes was analyzed with Image J software.
2.8Enzyme-linked immunosorbent assay (ELISA)ELISA was conducted strictly according to the instructions (R&D, USA). The OD value of each well was measured at a wavelength of 450nm within 15min.
2.9Nuclear and cytoplasmic extraction assayConducted according to the manufacture of NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo, USA).
2.10Statistical analysisData were exhibited as mean values ± standard error (mean ± SEM) of 3 independent experiments. Differences were evaluated by a two-tailed Student's t-test. Statistical significance was set at *P < 0.05, **P < 0.01, ***P < 0.001.
2.11Ethical StatementNot applicable.
3Results3.1HBx-overexpressing hepatoma cells differentially affect the angiogenesis of vascular endothelial cells varied on glucose concentration25mM glucose is the glucose concentration widely used in the culture of hepatoma cells. 30mM glucose is widely used as a high glucose condition in cellular investigations and 5.5mM glucose is analogous to normal blood sugar levels in humans. We displayed in Figure S1A that the hepatoma cells morphology is roughly the same between 25mM and 30mM glucose. Moreover, as shown with CCK8 analysis in Figure S1B, both in vector-transfected and HB-overexpressing hepatoma cells, 30mM glucose medium promoted the proliferation of hepatoma cells compared to 5.5mM glucose.
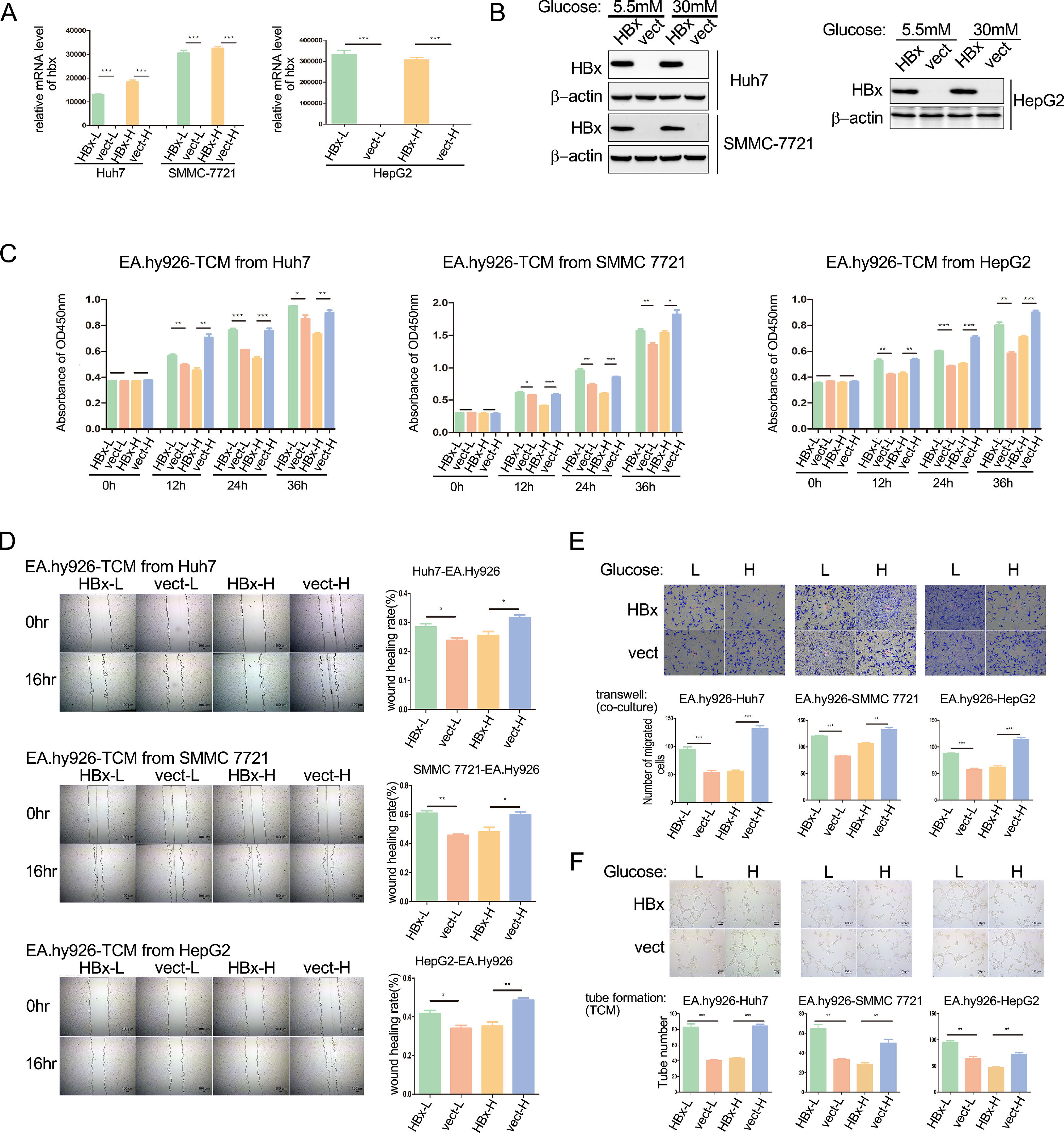
Our previous work has demonstrated that HBx alleviates the metastasis of hepatoma cells under a high glucose medium with 30 mM glucose while promoting it in a medium containing 5.5 mM glucose (to be published). We wondered whether HBx-overexpressing hepatoma cells play dual roles in HCC angiogenesis. The proliferation, migration, and tube formation of vascular endothelial cells determine the tumor angiogenesis ability. We transfected hbx plasmids to Huh7 or SMMC-7721 cells, and infected HepG2 cells with hbx adenovirus to build HBx-overexpressing cells. Analyzing with qPCR and western blot, we verified the overexpression of HBx proteins (Fig. 1A and B). Subsequently, supernatant medium in HBx-overexpressing hepatoma cells and vector control cells cultured in 30mM glucose (higher glucose concentration) or 5.5mM glucose (lower glucose concentration) respectively were collected as four kinds of tumor-conditioned mediums (TCM). As shown with CCK8 assays, compared to the vector control group, HBx-modified TCM from low glucose medium promoted the proliferation of EA.hy926 cells; meanwhile, HBx-modified TCM from high glucose inhibited the proliferation (Fig. 1C). Next, we conducted wound healing assays and illustrated that compared to TCM from the vector-transfected hepatoma cells, TCM collected from HBx-overexpressing hepatoma cells promoted the migration of EA.hy926 cells in the 5.5mM glucose medium, but significantly inhibited which in the 30mM glucose medium (Fig. 1D). Furthermore, we performed the transwell migration chamber experiments by co-culture of hepatoma cells in the lower chambers and EA.hy926 cells in the upper chambers. We observed that HBx-overexpressing hepatoma cells significantly promoted the migration of EA.hy926 cells in the 5.5mM glucose medium while inhibiting them in the 30mM glucose medium (Fig. 1E). Finally, we intervened EA.hy926 cells with different TCMs and performed tube formation assays. As shown in Fig. 1F, compared to the vector control group, the tube formation of EA.hy926 cells was significantly elevated after being cultured with TCM from HBx-overexpressing hepatoma cells in the 5.5mM glucose medium, while was weakened in the 30mM glucose medium. These results collectively demonstrated that the role of HBx-overexpressing hepatoma on angiogenesis varied by the different glucose conditions and inhibited the angiogenesis when in a high glucose condition.
 Q-PCR and western blot analysis of hbx overexpression in hbx-plasmids or hbx-adenovirus transfected hepatoma cells. Vect refers to vector. (C) CCK8 analysis of EA.hy926 incubated with tumor conditional medium (TCM). L refers to low glucose, and H refers to high glucose. (D) Wound healing analysis of EA.hy926 cells treated with TCM. (E) Transwell migration analysis of EA.hy926 co-cultured with hepatoma cells. (F) Tube formation analysis of EA.hy926 cells treated with TCM. *P < 0.05, **P < 0.01, ***P < 0.001. All of the histogram data in this figure were collected from three independent experiments.")
HBx-overexpressing hepatoma cells differentially affect the angiogenesis of vascular endothelial cells varying on glucose concentration. (A-B) Q-PCR and western blot analysis of hbx overexpression in hbx-plasmids or hbx-adenovirus transfected hepatoma cells. Vect refers to vector. (C) CCK8 analysis of EA.hy926 incubated with tumor conditional medium (TCM). L refers to low glucose, and H refers to high glucose. (D) Wound healing analysis of EA.hy926 cells treated with TCM. (E) Transwell migration analysis of EA.hy926 co-cultured with hepatoma cells. (F) Tube formation analysis of EA.hy926 cells treated with TCM. *P < 0.05, **P < 0.01, ***P < 0.001. All of the histogram data in this figure were collected from three independent experiments.
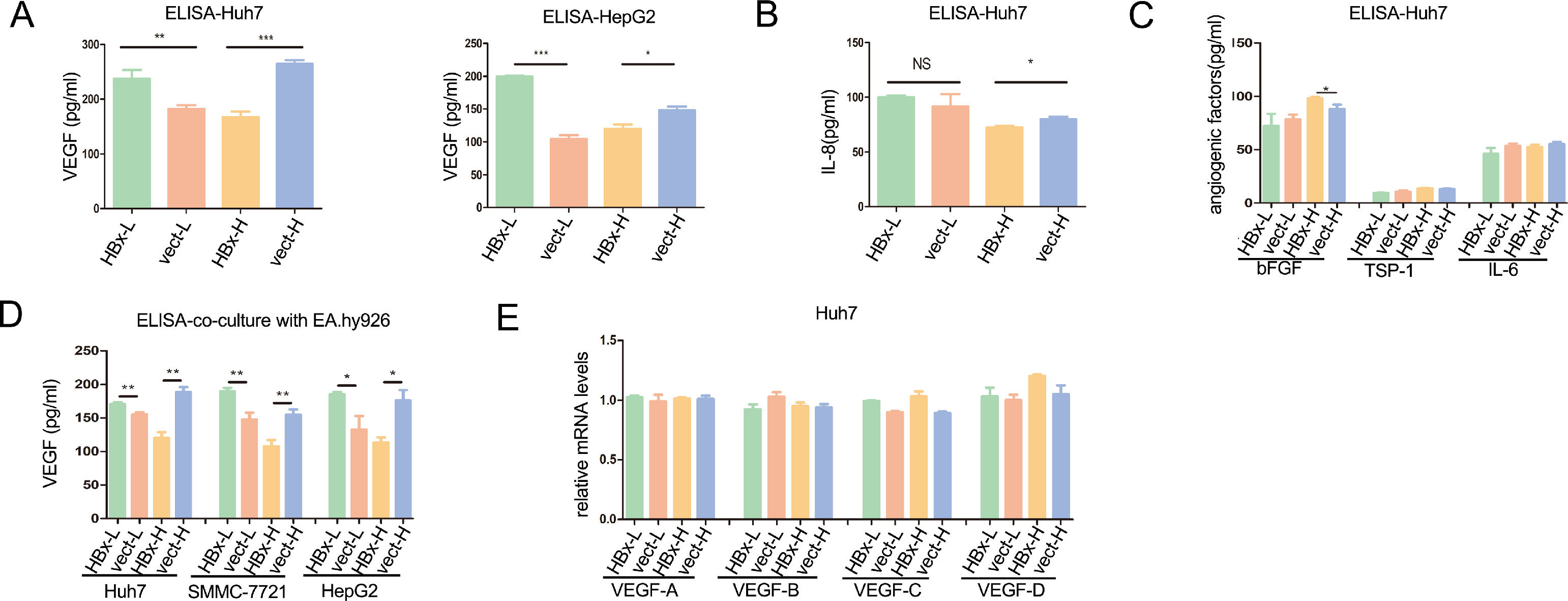
To explore how HBx-overexpressing hepatoma cells differentially regulated the angiogenesis of vascular endothelial cells, we detected the secretion levels of angiogenesis-related factors in HBx-overexpressing hepatoma cells. VEGF, also known as VEGF-A, VEGF is the key factor in triggering angiogenesis in endothelial cells [18]. As shown with enzyme-linked immunosorbent assay (ELISA), HBx significantly upregulated the secretion of VEGF both in Huh7 and HepG2 cells cultured with 5.5mM glucose medium, while significantly reducing the secretion of VEGF when cells incubated with 30mM glucose medium (Fig. 2A). Although VEGF being a major player in angiogenesis, tumor-induced angiogenesis is usually related to a complex interplay between multiple factors [2]. For the IL-8, an enhancer in endothelium permeability [19], HBx had no significant effect in regulating its secretion in Huh7 cells with 5.5mM glucose, while inhibiting the secretion in 30mM glucose (Fig. 2B). Additionally, basic fibroblast growth factor (bFGF) secretion, another pro-angiogenetic factor [20], was not altered by HBx in low glucose and slightly upregulated by HBx in high glucose (Fig. 2C). Additionally, HBx played no effect on the secretion of TPS-1 and IL-6 (Fig. 2C). We then co-cultured hepatoma cells with EA.hy926 cells and measured the concentration of VEGF in the medium. ELISA analysis came to the same results that all the VEGF expression levels were elevated when the three kinds of HBx-overexpressing hepatoma cells, Huh7, SMMC-7721, and HepG2, respectively being co-cultured with EA.hy926 in the 5.5mM glucose condition, compared to vector-transfected hepatoma cells, which were downregulated when in 30mM glucose condition(Fig. 2D). The VEGF family includes VEGF-A, VEGF-B, VEGF-C, VEGF-D [21]. Q-PCR analysis revealed that there was no difference in VEGF A-D mRNA levels between HBx-overexpressing and vector-transfected groups in all of the above interventions (Fig. 2E). These findings suggested that HBx in hepatoma cells played dual roles in regulating angiogenesis of vascular epithelial cells by differentially modulating the secretion of VEGF in hepatoma cells.
 ELISA analysis of VEGF and other angiogenic factors in supernatants of hepatoma cells. (D) ELISA analysis of extracellular fluid collected from the co-culture of EA.hy926 and hepatoma cells. (E) Q-PCR analysis of the VEGFs mRNA expressions in Huh7 cells. All of the histogram data in this figure were collected from three independent experiments.")
HBx plays dual roles in VEGF secretion from hepatoma cells according to glucose levels. (A-C) ELISA analysis of VEGF and other angiogenic factors in supernatants of hepatoma cells. (D) ELISA analysis of extracellular fluid collected from the co-culture of EA.hy926 and hepatoma cells. (E) Q-PCR analysis of the VEGFs mRNA expressions in Huh7 cells. All of the histogram data in this figure were collected from three independent experiments.
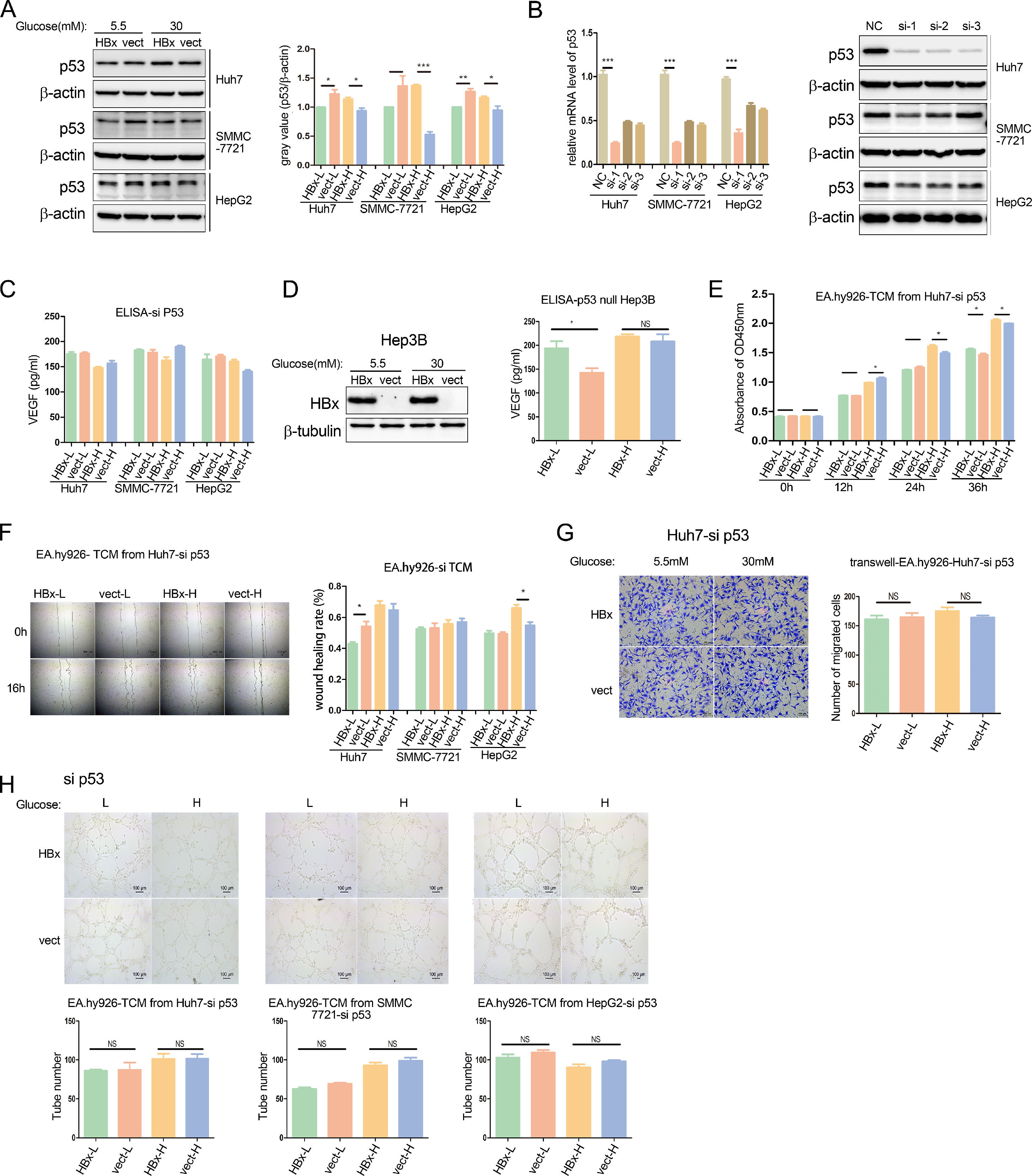
It's well documented that p53 negatively regulates VEGF expression inhibiting angiogenesis and metastasis [22]. We then analyzed the p53 protein expression in HBx-overexpressing hepatoma cells, compared to vector-transfected cells. Western blot results revealed that in the three hepatoma cell lines, HBx reduced p53 expression when cells were cultured in a 5.5mM glucose environment, while increased p53 abundance in the 30mM glucose Mellitus (Fig. 3A).
 Western blot analysis of p53 protein expression in HBx-overexpressing hepatoma cells. (B) Q-PCR and western blot analysis of p53 expression in hepatoma cells transfected with p53 siRNA. NC refers to negative control siRNA; si-1 refers to p53 siRNA 1; si-2 refers to p53 siRNA 2; si-3 refers to p53 siRNA 3. (C) ELISA analysis of VEGF secretion from hepatoma cells transfected with p53 siRNA. (D) Western blot analysis of HBx expression in Hep3B cells transfected with hbx-plasmids, and ELISA analysis of VEGF secretion in these Hep3B cells. (E, F, H) CCK8 analysis, wound healing analysis, and tube formation analysis of EA.hy926 treated with TCM from p53 siRNA transfected hepatoma cells respectively. (G) Transwell migration analysis of EA.hy926 co-cultured with p53 siRNA transfected Huh7 cells. All of the histogram data in this figure were collected from three independent experiments.")
HBx differentially regulates endothelial angiogenesis through the p53-VEGF axis. (A) Western blot analysis of p53 protein expression in HBx-overexpressing hepatoma cells. (B) Q-PCR and western blot analysis of p53 expression in hepatoma cells transfected with p53 siRNA. NC refers to negative control siRNA; si-1 refers to p53 siRNA 1; si-2 refers to p53 siRNA 2; si-3 refers to p53 siRNA 3. (C) ELISA analysis of VEGF secretion from hepatoma cells transfected with p53 siRNA. (D) Western blot analysis of HBx expression in Hep3B cells transfected with hbx-plasmids, and ELISA analysis of VEGF secretion in these Hep3B cells. (E, F, H) CCK8 analysis, wound healing analysis, and tube formation analysis of EA.hy926 treated with TCM from p53 siRNA transfected hepatoma cells respectively. (G) Transwell migration analysis of EA.hy926 co-cultured with p53 siRNA transfected Huh7 cells. All of the histogram data in this figure were collected from three independent experiments.
We further explored the role of the p53-VEGF axis in the HBx-involved tumor angiogenesis. Firstly, the silence of p53 expression was achieved by small RNA interference (siRNA) technology, and verified at the mRNA and protein levels respectively (Fig. 3B). In the p53 knockdown hepatoma cell lines, we found that the effects of HBx on regulating VEGF secretion levels disappeared in medium neither with 5.5mM glucose nor 30mM glucose. (Fig. 3C). In another p53-/- null hepatoma cell line, Hep3B, we transfected with hbx-plasmids, which overexpression was verified by western blot, and found that HBx also failed to inhibit VEGF secretion in the high glucose medium, excepting that it continued to promote VEGF secretion in the low glucose medium. (Fig. 3D). We presumed that HBx acted on vascular epithelial cells via extracellular fluids of hepatoma cells. We then collected a conditional medium from hepatoma cells with p53-knocked down (si-TCM) and examined the effect of the HBx-modified medium on vascular endothelial cells. CCK8 proliferation assays revealed that the HBx-mediated inhibition of EA.hy926 proliferation in the high glucose was reversed to a positive effect when the si-TCM intervention time was extended to 36hr, although the promotion effect of HBx in the low glucose was still the same (Fig. 3E). Furthermore, as evidenced by scratch tests, after the p53 expression was silenced in hepatoma cells, the HBx-involved regulation of EA.hy926 migration in low glucose or high glucose were either disappeared or unchanged varied on hepatoma cell lines. (Fig. 3F). In addition, we conducted a co-culture of EA.hy926 cells and p53-knocked down Huh7 cells in the migration chamber assay, and observed that there was no difference in the migration between HBx-transfected and vector-transfected groups neither in high glucose medium nor low glucose medium (Fig. 3G). These results illustrated that p53 knockdown tends to wipe out the role of HBx in modulating vascular epithelial cell proliferation and migration.
Lastly, we performed tube formation assays on EA.hy926 cells cultured with siTCM. We observed that the tube formations were no longer changed by HBx-modified medium (Fig. 3H). We demonstrated that p53 silence erased the effect of HBx on regulating vascular epithelial cell tube formation. In collection, these results revealed HBx acted on p53 to regulate VEGF secretion from hepatoma cells, and further modulate the angiogenesis in vascular epithelial cells.
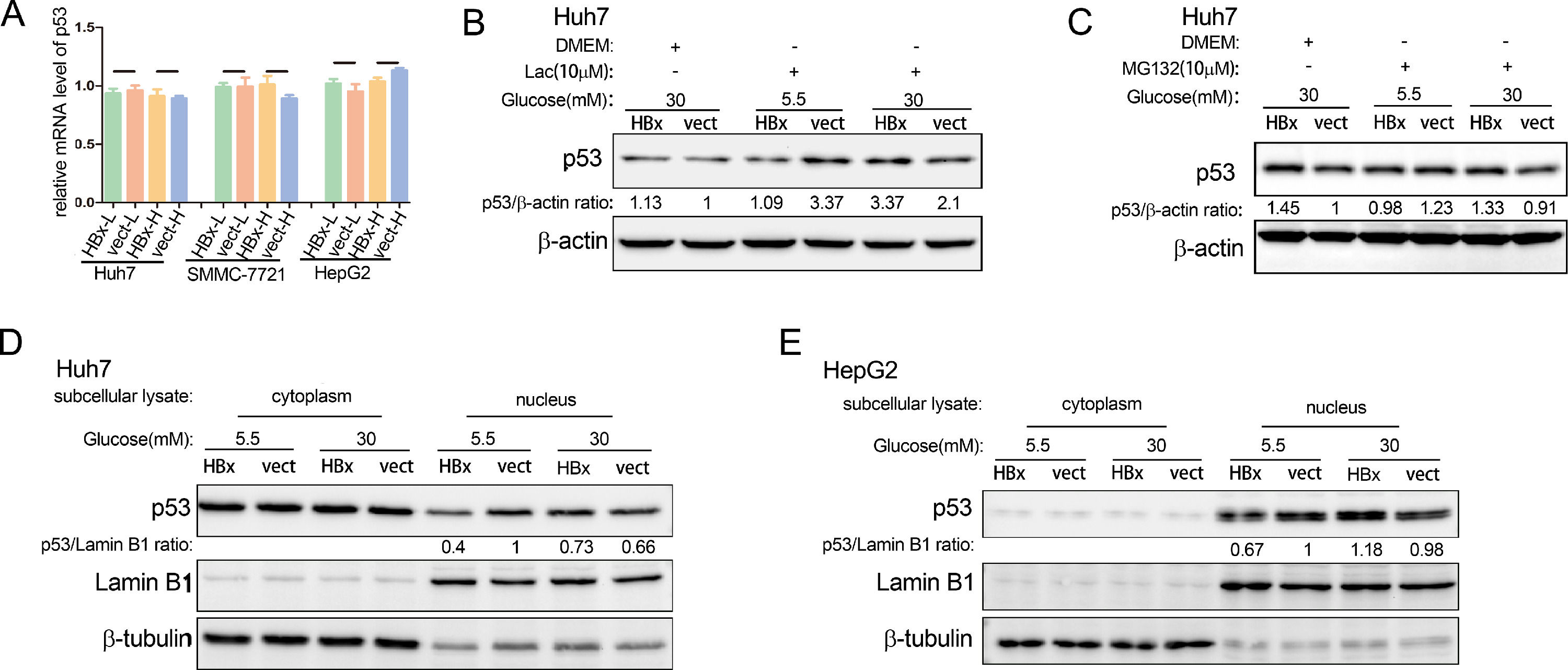
3.4The way HBx regulates p53 is complex and mainly in the nucleusWe then explored the ways of HBx in regulating p53 expression. Using qPCR, we found that no matter whether in high glucose medium or low glucose environment, HBx did not affect the p53 mRNA expression in hepatoma cells (Fig. 4A). It's documented that the p53 protein is degraded by the ubiquitin-proteasome pathway [23]. We showed that lactacystin, a selective 20S proteasome inhibitor, increased the p53 abundance in hepatoma cells, verifying the involvement of ubiquitination proteasome pathway in p53 protein degradation (Fig. 4B). However, neither inhibitor of the proteasome degradation pathway, MG132 or Lactacystin, changed the regulatory trends of HBx on p53 protein levels (Fig. 4B and C). These results revealed that the level of p53 protein in hepatoma cells regulated by HBx under different glucose conditions is neither related to transcriptional regulation nor ubiquitination proteasome pathway.
 Q-PCR analysis of p53 mRNA expression in HBx-overexpressing hepatoma cells. The Q-PCR data were collected from three independent experiments. (B-C) Western blot analysis of p53 protein abundance in HBx-overexpressing hepatoma cells treated with lactacystin (Lac) or MG132. (D-E) Western blot analysis of cytoplasmic and nuclear p53 protein expression in hepatoma cells. Lamin B1 serves as the loading control of nuclear protein; b-tubulin serves as the loading control of cytoplasmic protein.")
The way HBx regulates p53 is complex and mainly in the nucleus. (A) Q-PCR analysis of p53 mRNA expression in HBx-overexpressing hepatoma cells. The Q-PCR data were collected from three independent experiments. (B-C) Western blot analysis of p53 protein abundance in HBx-overexpressing hepatoma cells treated with lactacystin (Lac) or MG132. (D-E) Western blot analysis of cytoplasmic and nuclear p53 protein expression in hepatoma cells. Lamin B1 serves as the loading control of nuclear protein; b-tubulin serves as the loading control of cytoplasmic protein.
P53 is distributed in diverse subcellular organelles. We extracted cytoplasmic and nuclear protein lysis respectively and found that HBx reduced the nuclear p53 protein abundance in the low glucose and increased it in the high glucose in Huh7 cells (Fig. 4 D). In the meanwhile, HBx had little effect on the p53 protein abundance in the cytoplasm (Fig. 4D). Similar results were observed in HepG2 cells (Fig. 4E). These results demonstrated that HBx mainly regulated the p53 protein in the nucleus.
4DiscussionHBx is critical for the activation of HCC angiogenesis. Glucose concentration differs in zones of liver tumor interstitium. We here show that HBx plays dual roles in HCC angiogenesis according to the glucose levels in the milieu. In low glucose, HBx-overexpressing hepatoma cells reduced the abundance of nuclear p53 contributing to the subsequently increased paracrine of VEGF-a to vascular endothelial cells, promoting the proliferation, migration, and tube formation of endothelial cells. However, when in the high glucose condition, the phenomenon is converse.
Another report has characterized the glucose-deprived condition in liver tumor interstitial, illustrating that the glucose level was approximately 0.8mM, compared to the 15mM glucose in the normal liver [24]. Concerning the glucose shortage condition, it's well understood that HBx was widely characterized to increase the malignant potential of HCC in vivo. However, there are limited approaches to measuring local glucose concentrations in vivo [15]. It cannot be ignored that the uneven distribution of glucose within a bulk tumor, the elevated glucose concentration in the diabetes milieu, and the high concentration of glucose diffusing from the portal vein to the sinusoid just absorbed from the intestine. It has been reported that high glucose-induced c‑Met activation enhanced aggressive phenotype in HCC cells [25]. We demonstrated that HBx along with high glucose-supplemented conditions surprisingly inhibited HCC angiogenesis. HBx may alter the supposed oncogenic role of high glucose. Furthermore, we illustrated that HBx plays the opposite role in HCC angiogenesis in high glucose versus low glucose, through differentially modulating the secretion of VEGF from hepatoma cells. Considering the mix of VEGF signals from different hepatoma cells in the extracellular milieu in vivo may explain why the dual effects of HBx are usually masked, which requires future work with animal models and patients.
Neovascularization provides nutrients for tumor growth and leaky vessels that facilitate the intravasation of tumor cells [3,26]. The dual roles of HBx in regulating angiogenesis make hepatoma cells escape from a nutritionally deficient environment and stationed in places rich in glucose, benefiting the development of HCC as a whole. Consisting with the notion that VEGF is the key mediator of HCC angiogenesis [4,27], our data suggested that HBx targets VEGF to regulate HCC angiogenesis. Although HBx is a transcription factor, all of the current papers, including our study, have demonstrated that HBx indirectly regulates VEGF expression by activating signal transduction or other nuclear factors [28]. Unlike previously reported regulation of VEGF mRNA levels by HIF1a [29], our results suggest that HBx and p53 regulate VEGF secretion levels but have no effect on VEGF mRNA levels. Our data suggest that VEGF serves effectively as a communicator between HBV-infected hepatoma cells and stroma cells, and HBx regulates intercellular communication to accelerate HCC progression.
We identified that p53 was required for the dual role of HBx on HCC angiogenesis. The HBx-induced angiogenic phenotype disappeared in p53-null Hep3B cells and p53 siRNA-treated hepatoma cells. P53 is a well-characterized tumor suppressor in HCC. In liver cancer patients, the mutation and loss of the p53 gene in hepatocellular carcinoma cells happens sometimes [30,31]. Similarly, it's reported that dual effects of HBx on the G1–S checkpoint control for cell-cycle depend on whether functional p53 is maintained [32]. HBx prolonged arrest in G1 leads to apoptosis when p53 is present; In the meanwhile, in the absence of p53, HBx induces rapid and uncontrolled cell proliferation [32]. It can be inferred that in vivo situations, the inhibition of HCC angiogenesis by HBx together with high glucose sometimes may not occur.
Our data suggest that HBx regulates HCC angiogenesis through the p53-VEGF axis. The wild-type p53 plays dual roles in the regulation of VEGF promoters in that it can positively regulate VEGF expression in the initial phases of hypoxia, and also indirectly repress VEGF expression under continued hypoxic challenge [22]. Consistent with our data, it's reported that silencing the p53 gene in hepatoma cells (with WTp53) induces VEGF expression and inhibits the migration of endothelial cells [33]. As an important transcription factor, we speculate that p53 nuclear changes may further affect the transcription of angiogenesis-related factors. However, through Q-PCR detection of VEGF mRNA, the transcription level changes have not yet been identified. The way p53 regulates VEGF secretion needs further exploration.
Mickle studies are questing the interaction of HBx and p53. It's reported that through protein-protein interaction [34], HBx sequesters p53 to the cytoplasm of hepatocytes, partially preventing the p53’s nuclear entry and inhibiting several critical p53-mediated cellular processes [35]. We observed that HBx differentially regulated nuclear p53 protein abundance, without affecting the mRNA expression. Although the p53 protein was documented to be degraded by the ubiquitin-proteasome pathway [23], our experiments proved this pathway was not involved in the program of HBx-regulated p53 expression. Recently, ubiquitin-independent p53 proteasomal degradation has been proposed [36]. Therefore, we infer that regulation in p53 mRNA editing or other protein degradation pathways may participate in the HBx-p53 process, which needs further investigation.
5ConclusionsTaken together, HBx elevates nuclear p53 protein expression to block VEGF secretion in hepatoma cells, leading to alleviated angiogenesis in high glucose; which is reversed in low glucose. We suggest that the dual roles of HBx confer hepatoma cells to remain in a glucose-rich environment and escape from the glucose-low milieu through tumor vessels. The dual roles of HBx in regulating HCC angiogenesis provide a survival advantage for the HBx-expressing hepatocytes in liver carcinogenesis, and may promote the development of HCC in general.
Authors contributionsGuitao Xiao, acquisition of most of the data, analysis, and interpretation of data; Xiaoyun Huang, acquisition of some of the data, drafting of the manuscript, study concept and design, obtained funding; Tingxuan Huang, analysis and interpretation of data, statistical analysis; Zhixin Chen, statistical analysis, administrative, technical, or material support; Yuehong Huang, administrative, technical, or material support; Rongfeng Huang, critical revision of the manuscript for important intellectual content, obtained funding, study supervision; Xiaozhong Wang, study concept and design, critical revision of the manuscript for important intellectual content, study supervision.
Data availability statementDerived data supporting the findings of this study are available from the corresponding authors on request.
This work was supported by Startup Fund for Scientific Research, Fujian Medical University, China [grant number 2019QH1037]; Fujian Natural Science Foundation, China [grant number 2021J05048]; Fujian Provincial Science and Technology Innovation Joint Fund Project, China [grant number 2020Y9093].




{kind=link}