




Hepatitis C virus (HCV) is a small, enveloped RNA virus. The number of HCV-infected individuals worldwide is estimated to be approximately 200 million. The vast majority of HCV infections persist, with up to 80% of all cases leading to chronic hepatitis associated with liver fibrosis, cirrhosis, and hepatocellular carcinoma. The interaction between HCV and the host have a pivotal role in viral fitness, persistence, pathogenicity, and disease progression. The control of HCV infection requires both effective innate and adaptive immune responses. The HCV clearance during acute infection is associated with an early induction of the innate and a delayed initiation of the adaptive immune responses. However, in the vast majority of acute HCV infections, these responses are overcome and the virus persistence almost inexorably occurs. Recently, several host- and virus-related mechanisms responsible for the failure of both the innate and the adaptive immune responses have been recognized. Among the latter, the wide range of escape mutations to evade the specific-T-and B-cell responses as well as the T cell anergy and the CD8+ T cell exhaustion together with the interference with its function after prolonged virus exposure hold a pivotal role. Other HCV strategies include the modification or manipulation of molecules playing key roles in the induction of the interferon response and its induced effector proteins. In this review, we attempt to gain insights on the main T cell immune evasion strategies used by the virus in order to favor its persistence.
Hepatitis C virus (HCV) infection represents the most common blood-borne viral infection for which a vaccine is still lacking, while an estimated 3% of the world’s population is infected, representing approximately 200 million individuals.1 The control of the acute infection depends on both host- and viral-related factors. The former includes both an early induction of innate- and a delayed initiation of adaptive-immune responses. However, in the majority of acutely HCV infected individuals, these responses are insufficient to clear the virus and most of HCV infections persists, with up to 80% (50–90%) of all cases leading to chronic hepatitis, eventually associated with liver fibrosis, cirrhosis and/or hepatocellular carcinoma. The acute phase of infection is usually asymptomatic, with rare cases exhibiting fatigue, malaise and jaundice during a few weeks. Viral RNA can be detected 1-3 weeks after viral entry into the organism, despite a high induction level of the innate immune response.2 Intriguingly, HCV presence seems to be virtually overlooked by both T cell and B cell adaptive immune responses during the first 8-12 weeks after infection (e.g., antibodies to HCV may be evident by 3 months after infection, in approximately 90% of infected individuals).3 An elevated level of alanine-aminotransferase (ALT) within 4-12 weeks after exposure is an indication of liver cell injury. Immune events during this stage of disease are considered to be essential for determining the outcome of infection.
Interferon Lambda (IFN-λ):An Essential Partner of The T Cell ResponseAlthough the impact of both interferon stimulated genes (ISG) and IL-28B (a subclass of IFN-λ, a type III IFN) on the course of HCV natural infection are well beyond the scope of this concise review, a brief comment about the latter is well deserved, considering its current prognostic usefulness for clinicians and its connection with T cell response. IFN-λ is the most recently group of identified IFN proteins, which includes IFN-λ1, -λ2, -λ3 and -λ4, encoded by IFNL1 (formerly, IL-29), IFNL2 (IL-28A), and IFNL3 (IL28B) and IFNL4 genes, respectively. In humans, IFNL1, IFNL2 and IFNL3 are functional genes, while IFNL4 is a pseudogene in part of the population due to the single nucleotide polymorphisms (SNP) rs368234815.4
Viruses can be sensed in infected cells through the interaction of pathogen-associated molecular patterns (PAMPs) with sensors such as cytosolic helicases (e.g. RIG-1, MDA-5 or LGP-2), membrane-associated Toll-like receptors (TLRs) and Nod-like receptors (NLRs). After engaging with their respective ligands, these pattern recognition receptors activate IKKe/TANK-binding kinase 1 (TBK-1), the IKKa/β/γ complex, as well as MAPK, leading to the activation of transcription factors, such as IFN regulatory factor 3 (IRF-3), IRF-7, NF-κB, and AP-1. Once activated, these factors translocate into the nucleus and, in turn, activate the transcription of type I IFN. In an autocrine as well as a paracrine way, the IFN-α/β activates the expression of ISGs through the JAK-STAT signaling pathway to establish an antiviral state in universally distributed host cells. Likewise, IFN-λs are believed to activate the same pathway promoting the expression of a set of antiviral proteins in epithelial cells including hepatocytes and in as yet not properly defined hematopoietic cells.4
Indeed, both the T cell response as well as the SNPs in the proximity of the IFNL3 gene appears to be crucial for viral clearance.4,5 The molecular mechanism underlying the association between SNPs and the outcome of natural clearance of HCV or the response to IFN therapy in patients chronically infected with HCV has been proposed recently by McFarland, et al. They have identified a favorable SNP located in the 3’ untranslated region of IFNL3 mRNA that diminishes the binding of HCV-induced microRNAs (miR-208b and miR-499a-5p) during infection. Thus, this SNP dictates transcript stability and influences the outcome of both antiviral treatment and spontaneous clearance during HCV infection.6 Additionally, a variant upstream of IFNL3 creating a new interferon gene –referred as the ancestral ‘AG’ allele of IFNL4– is associated with impaired clearance of hepatitis C virus. This new genotype results in the expression of four previously unknown proteins, including p179, designated IF-Ν-λ4. This cytokine induces a weak cellular signaling that may reduce the responsiveness of cells to IFN-α, thereby inhibiting efficient HCV clearance.7 In sharp contrast, the TT allele of such SNP disrupts the open reading frame of IFNL4 (thus, inhibiting its translation) and protects against HCV. These findings suggest that IFN-λ4 is the causative agent of HCV clearance failure. Remarkably, the very recent discovery of an additional SNP (rs117648444) acting in combination with the AG SNP proved also useful to predict the response to anti-HCV treatment.8
The reader is kindly prompted to look at both figures 1 and 2, as well as to table 1 to follow the key concepts depicted in this concise review.
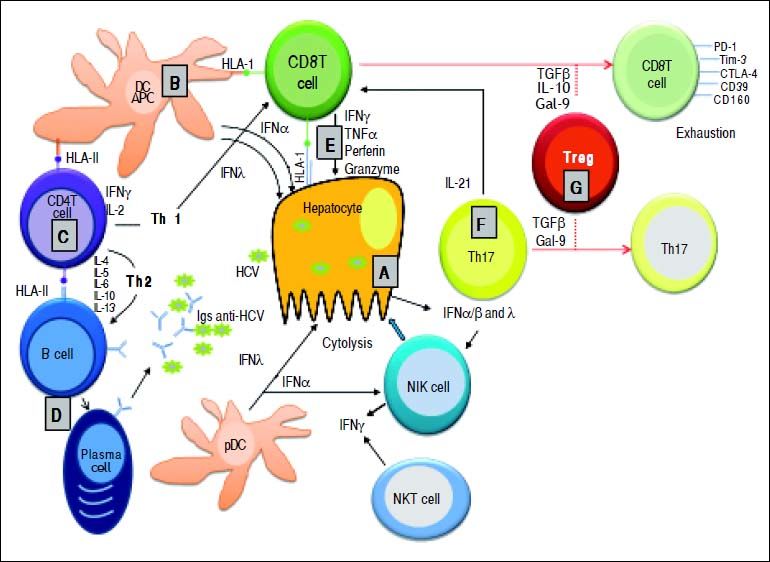
 and myeloid (m) dendritic cells (DCs). These cytokines activate NK cells for killing HCV-infected hepatocytes. B. Küpffer cells (as liver antigen-presenting cells) and DCs present HCV-derived epitopes to both CD4+ and CD8+ T cells in the context of HLA class II and HLA class I, respectively. C. CD4+ helper T cells activate both CD8+ T cells and B cells through production of Th1 (IFN-γ IL-2) and Th2 (IL-4, IL-5, IL-6) cytokines, respectively. D. Anti-HCV specific Igs are released from plasma cells during primary infection. E. Clearance mechanisms are mediated by CD8+ T cells exerted by cytolytic (perforin and granzymes), or non-cytolytic mechanisms through secretion of IFN-γ and TNF-α. F. IL-21 (mainly produced by Th17 cells, but also by NKT cells) sustains CD8+ T cell functions and is able to rescue virus-specific Tcells from exhaustion. G. Regulatory T cells might decrease primary immune response by secreting the regulatory cytokines TGF-β, IL-10, or by expressing Gal-9 that enhances apoptosis of Tim-3+ CD4+ and CD8+ T cells.")
Essential features of the innate and adaptive immune response during primary HCV infection. A. HCV replication in hepatocytes promoting the release of type I and type III IFNs. IFN-α and - λ are also secreted by both plasmocytoid (p) and myeloid (m) dendritic cells (DCs). These cytokines activate NK cells for killing HCV-infected hepatocytes. B. Küpffer cells (as liver antigen-presenting cells) and DCs present HCV-derived epitopes to both CD4+ and CD8+ T cells in the context of HLA class II and HLA class I, respectively. C. CD4+ helper T cells activate both CD8+ T cells and B cells through production of Th1 (IFN-γ IL-2) and Th2 (IL-4, IL-5, IL-6) cytokines, respectively. D. Anti-HCV specific Igs are released from plasma cells during primary infection. E. Clearance mechanisms are mediated by CD8+ T cells exerted by cytolytic (perforin and granzymes), or non-cytolytic mechanisms through secretion of IFN-γ and TNF-α. F. IL-21 (mainly produced by Th17 cells, but also by NKT cells) sustains CD8+ T cell functions and is able to rescue virus-specific Tcells from exhaustion. G. Regulatory T cells might decrease primary immune response by secreting the regulatory cytokines TGF-β, IL-10, or by expressing Gal-9 that enhances apoptosis of Tim-3+ CD4+ and CD8+ T cells.

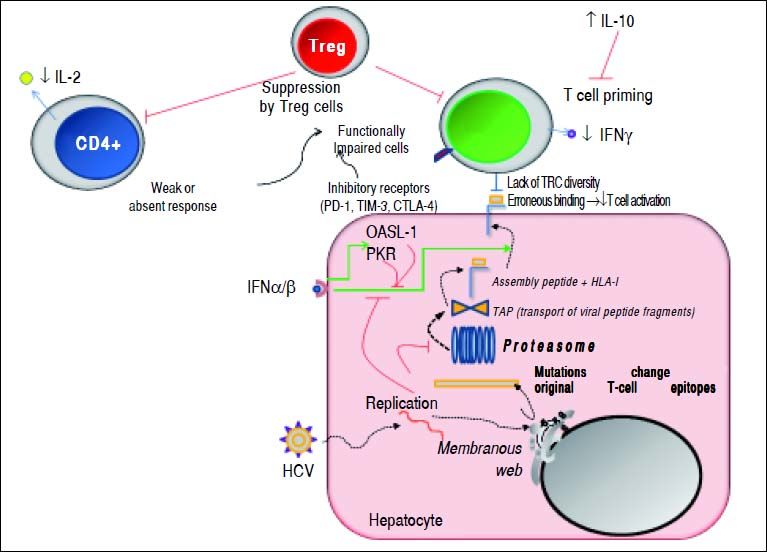
Mechanisms used by HCV to escape from adaptive cellular immune response. During viral replication, HCV proteins are generated; then, these molecules are degraded by the proteasome complex to generate peptide fragments that are translocated to the ER in order to assembly with HLA-I molecules. These complexes are transported to the hepatocyte surface to be presented to CD8+ T cells. Once activated, these cells secrete IFN-γ. The secreted levels of both IL-2 and IFN-γ by CD4+ and CD8+ T cells are respectively diminished. IL-10 is able to suppress naïve HCV-specific CD8+ T cell priming. Red lines indicate strategies to suppress the immune response. Dotted lines show the steps throughout the HCV replication as well as HCV-derived viral epitopes presentation on hepatocyte surface by HLA-I. The green lines denote the stimulatory effect of type I IFNs on HLA-I expression on cell surface and on both PKR and OASL-1 molecules. For further detals, see the man text.
Highlights on HCV strategies for viral evasion from the adaptive cellular immune response.
| Viral strategies | References | |
|---|---|---|
| Escape mutations to evade the specific-T cell response | -Proteasomal processing (mutations in the flanking region)-Binding to HLA molecules (mutations at the HLA binding anchors)-TCR recognition (mutations in the centre of the epitope) | 7 |
| Original antigenic sin | 27 | |
| Clustered accumulated mutations | 24 | |
| Inhibitory signal to HCV-specific CD8+ T cells | 33, 35, 36 | |
| T cells primed improperly became anergic | 25, 30 | |
| T cells exhausted under a prolonged and excessive stimulation | 32, 37, 38 | |
| CD8+ T cell exhaustion after prolonged virus exposure | Expression of inhibitory receptors (PD-1, Tim-3, 2B4, KLRG-1, CTLA-4, and CD160), or their ligands in liver cells (PDL-1, Gal-9 and other molecules) | 22 |
| Expansion of Tregs | 46, 50, 51 | |
| CD4+ T cell response failure | Decrease of IL-21producing Th17 cells and increase of Gal-9 producing Foxp3+ regulatory CD4+ T cells | 22 |
| Failure of IL-2 secretion and disruption of IFN-γ and cellular proliferation | 44 |
The outcome of HCV infection is mainly influenced by the coordinated action of heterogeneous cell types. In this regard, numerous reports provide evidence that the HCV clearance depends on the magnitude, breadth and quality of the virus-specific CD4+ and CD8+ T cell responses to multiple epitopes derived from both structural and non-structural proteins.9 For the first time, and as a consequence of the delayed induction -but not to a cellular recruitment failure-, such immune responses are detected in blood 4 to 8 weeks after infection.10 The primary effector response is mediated by CD8+ T cells, whose function is sustained by the CD4+ T cells, both of which are responsible for long-lasting control of the HCV.11 After the onset of the HCV-specific CD8+ T cell response, a direct relationship is established with the HCV clearance. A vigorous response related to viral clearance and targeting multiple epitopes is detected in T cells recovered either from the liver or from peripheral blood. Such antiviral activity is mainly exerted through IFN-γ-mediated non-cytolytic effector functions and merely to a lower extent through cytolytic effector functions, exerted by perforin as well as by Fas/FasL interactions. The non-cytolytic antiviral activity is 100- to 1,000-fold more effective than the cytolytic one.12 However, other CD8+ T cell functions with a protective role are also observed, such as the production of IL-17 from a subset of cells able to recognize dissimilar epitopes than those targeted by IFN-γ secreting CD8+ T cells.13
From the host, the HCV-specific CD8+ T cell response is influenced by the HLA class I alleles. HLA class I alleles A*0301, B*2701, B*5701, B*5703 and C*0102 are associated with viral clearance, whereas B8 is linked to viral persistence. The strongest protective effect is observed with HLA-B27.14 Remarkably, HLA class I expression is up-regulated by both type-I and -II IFNs.
The HCV-specific CD4+ T cells are critical for both limiting immune evasion and priming effector memory CD8+ T cells. Interestingly, among acute HCV patients and irrespectively of their clinical outcome- primed broad HCV-specific CD4+ T cell responses directed against multiple epitopes are observed in blood, which persist for long periods in patients that spontaneously resolve the infection. This event almost exclusively takes place during the first year of infection -and usually within the first 6 months-, stressing the fact that the virus tends to gain the battle against the host as time elapses. Moreover, when the infection evolves to persistence, the HCV-specific T cell responses are not sustained and become specifically dysfunctional for HCV.15 In the latter scenario, virus and host cohabit avoiding accelerated liver damage. Similarly to the findings described for HLA class I, certain HLA class II alleles influence the outcome of HCV infection as well. The HLA class II alleles most reproducibly associated with viral clearance are DRB1*1101 and DQB1*0301, which are genetically close-linked. However, HCV-derived CD4+ T cell epitopes showed promiscuity and can be presented by different HLA II molecules.16 As previously described for CD8+ T cells, IL-17-producing CD4+ T cells were observed among chronic infected patients, but their role in the outcome is still debated. These T cells displayed a distinct phenotypic profile, high expression of the homing receptor CD161 and low levels of inhibitory receptors, such as mucin-domain-containing-molecule-3 (Tim-3) and programmeddeath 1 (PD-1).17
Escape Mutations to Evade the Specific-T Cell ResponseWhether the HCV escape from T cell response is a cause or a consequence of HCV persistence is still under extensive debate. However, several important insights into CD8+ T cell-mediated viral escape have been revealed. The HCV exhibits a proclivity to mutate due to its high rate of replication (up to 1012–14 particles/day or about 106 IU/mL of plasma) associated with a lack of proofreading ability of its polymerase. Therefore, multiple virus variants or quasispecies co-circulate in an individual patient facilitating the selection of CD8+ T-cell escape variants. Hence, their occurrence responds to a complex interplay of the host T cell responses, the viral epitopes evolution under immune selection pressure and the viral fitness costs, although they may be restricted due to the incompatibility with an abolished viral replicative capacity. Four distinct outcomes may take place, as depicted in the following paragraph:
- •
The viral clearance during acute infection is associated with a sustained CD4+ T cell response with multi-specific CD8+ T cell response that may constrain the development of viral escape mutations. Remarkably, the early CD4+ T cell responses tend to disappear from the blood in those patients who develop HCV persistence.18 This failure of the CD4+ T cell response during the acute phase of the HCV infection may be due to a decrease in IL-21-producing Th17 cells and an increase of galectin-9 (Gal-9) producing Foxp3+ regulatory CD4+ T cells.19
- •
The above mentioned HCV escape mutations do not likely develop when the CD8+ T cell response is weak, associated with an absent or impaired CD4-help and priming. In this scenario, high viral levels and intact HCV epitopes are associated with increased levels of T cell inhibitory receptors.
- •
In contrast, when a discrete failure of the CD4+ T cell response occurs in the presence of a dysfunctional narrow but vigorous CD8+ T cell response, the appearance of escape mutations is favored and additional compensatory mutations may be required for replicative fitness improvement. These facts are associated with a decreased inhibitory receptor expression on CD8+ T cells, perhaps accounting for a HCV robust proliferation to wild-type, non-mutated virus.
- •
Finally, without both a restricting HLA allele and a CD8+immune selective pressure, reversion to the wild-type sequence probably occurs as a consequence of the high fitness cost associated with some escape mutations.9 These HCV mutations may occur at 3 distinct sites regarding T cell epitopes:
- a)
In a flanking region of these molecules, leading to a proteasomal processing of an abnormal peptide, which may be cleaved within the epitope and hence, without being loaded onto the HLA-I molecule.
- b)
Within the HLA-I anchor site (i.e. P2 and the C-terminal amino acid which fails to promote CD8+ T cell activation), or
- c)
Within T cell binding site (i.e. in the center of the epitope, which prevents a proper activation of CD8+ T cells as well or even inducing it as an antagonist).9,20
- a)
The HLA-I allele background could also play an important role in determining viral escape, since it has been reported a strong association between specific HLA-I alleles and viral escape within an immunodominant HLA-re-stricted epitope (e.g. HLA-B27). Importantly, the occurrence of clustered accumulated mutations within the epitope is required for efficient CD8+ T-cell escape despite the broad cross-recognition of viral variants. Thus, immune escape probably requires a too long elapsed time, providing enough time to the host immune system to clear the virus before escape can take place.21 These strategies constitute a means to escape HLA-restricted immune responses but considering the HCV fitness costs, such ability is constrained and compensatory mutations may be required to restore HCV replicative capacity.22 When the mutated epitopes are no longer bound by the HLA-I molecule, no substantial additional diversification is expected. However, those epitopes that still have the capacity to bind to the HLA molecule may trigger the “original antigenic sin” phenomenon that results in a diminished clearance of viral variants and therefore, enhancement of viral escape. In this regard, two pathways might take place:
- •
The expansion of wild-type specific CD8+ T cells as a consequence of stimulation by mutated epitopes23 and
- •
Alternatively, the inhibition of HCV-specific CD8+ T cells responding to HLA-I mutated epitopes after TCR recognition due to inhibitory signaling. Despite its elevated frequency among targeted CD8+ T-cell epitopes, viral escape mutations are not a general consequence resulting from CD8+ T-cell pressure. In this regard, a large proportion of intrahepatic virus-specific CD8+ T-cells was detected to target the non-mutated viral antigens among chronically infected patients. Thus, it suggests that factors other than mutational escape contribute to the failure of intrahepatic virus-specific CD8+ T-cells.20
Regarding the HLA class II-restricted epitopes escape mutations can also occur, but they are infrequent among chronically infected patients.24 Considering both HLA-I and -II, it is plausible to infer that additional mechanisms could contribute to CD8+ and CD4+ T-cell failure, such as T-cell dysfunction. In this context, two distinguishable situations are possible in chronic HCV infection. By the one hand when T cells are improperly primed by signaling through the TCR in the absence of costimulatory or inflammatory signals, they become anergic.25,26 By the other hand, when T cells become exhausted and primed by antigen, as well as costimulatory and inflammatory signals, they still develop some effector functions. But then, under a prolonged and excessive stimulation, progressive loss of functions occurs.26
CD8+ AND CD4+ T Cell Function ImpairmentViral persistence is prompted when CD8+ T cell exhaustion emerges. The prolonged exposure appears to be the mechanism that leads to T cell dysfunction in chronic hepatitis C. Such persistent high level of viremia is firstly accompanied by a good T cell response but then it fails to eliminate the virus with a subsequent gradual decline of CD8+ and CD4+ T cell responses. Two main pathways have been described that mediate this exhaustion:
- •
The expression of inhibitory receptors on CD8+ T cells, and
- •
The expansion of regulatory T cells (Tregs ) that suppress CD8+ T cell activity.
The former involves changes in cytokines’ levels production or cell proliferation after antigen-dependent stimulation that prime T-cells for activation-induced apoptosis.27 Such T-cell exhaustion is sustained by the over-expression of inhibitory receptors like Tim-3, PD-1, cytotoxic T-lymphocyte antigen 4 (CTLA-4), CD160 and 2B4 on HCV-specific CD8+ T cells in the blood and liver of individuals developing chronic HCV infection. Likewise, the expression of their respective ligands in the liver may contribute to exhaustion and apoptosis, as occurs when the PD-1 ligand-1 (PDL-1) and the Tim-3 ligand Gal-9 are highly expressed.19 Amazingly, a HLA-I restricted CD8+ T cell -to-CD4+ T cell cross-differentiation might occur with the same clonotypic T cell receptor. This conversion could produce HLA-class-I-restricted CD4+Foxp3+ Treg cells,28 whose implications still remain to be explored in HCV infection. The dysfunctional HCV-specific CD8+ T cells also exhibit a low interleukin-7 receptor (CD127) expression, able to regulate CTL reactivity through the balance modulation between Mcl-1(myeloid leukemia cell differentiation protein-1) and Bim (Bcl2-interacting mediator). Bim is a pro-apoptotic molecule blocked by the action of Mcl-1. A low exvivo Mcl-1 expression and Bim up-regulation after antigen encounter were involved in CD127(low) HCV-specific CTL hyporeactivity during chronic infection.29,30 While this manuscript was under review, terminally exhausted CD8+ T cells have been identified as CD39+ cells.31
The T cell exhaustion has been proposed as a mechanism underlying the dysfunction of HCV-specific CD4+ and CD8+ T cells during acute infection. However, this proposal is still under intense debate. If true, it should indicate that the fate of the HCV infection is already determined at its very early stage. In this regard, at least four groups initially observed contrasting results, since high PD-1 levels on HCV-specific T cells during acute infection were observed to be associated with viral persistence, requiring preservation of cognate antigen during the chronic stage,32 while such high PD-1 level on both HCV-specific CD8+ and CD4+ T cells during acute HCV infection, appeared to be unrelated to the clinical outcome in other studies.33 Perhaps, a conciliating view might take into account that T cell exhaustion is a dynamic process which exhibits variable stages of impairment, as observed in the experimental lymphocytic choriomeningitis virus (LCMV) model,34 and confirmed by elegant ex vivo experiments on acute HCV patients who either spontaneously resolved the infection or became persistently infected, pointing to crucial and distinct roles of Tim-3 and PD-1 molecules.32 Importantly, the frequency of the memory precursor marker CD127 on HCV specific T cells has been recently proposed to predict the outcome of the HCV acute infection in experimentally infected chimpanzees, since high levels appeared to be present only in those who cleared the infection.35 The targeting of multiple co-regulatory receptors has very recently opened new avenues for therapeutic approaches.36
Several groups have demonstrated weak or absent HCV-specific CD4+ T cell responses during chronic HCV infection with a failure of IL-2 secretion that may lead to disruption of IFN-γ and cellular proliferation, as opposed to physical deletion or complete functional unresponsiveness. Such inhibition of IL-2 secretion -but not of its transcription-has also been recently associated with the interaction of HCV E2 glycoprotein with T cells.37 After a decreased production of IL-2, the cytotoxicity and production of TNF-α and IFN-γ are impaired sequentially. The recent discovery of CD154 molecule (CD40L) as a biomarker of antigen specific activated CD4+T cells might help to overcome some of the drawbacks of functional assays and limitations of multimer-based methods used for the detection of this crucial population,38 thus answering a key question: is this a matter of an insufficient number of activated CD4+ T cells and/or a result of functional impairment? In this regard, it should be stressed that HCV is able to infect T cells. Interestingly, HCV E2 RNA as well as E2 protein are independently able to suppress either proximal and distal TCR signaling, respectively. Most likely both structures are involved in HCV pathogenesis, contributing to viral persistence.39 Moreover, HCV F protein promotes the PD1 expression on both CD8+ and CD4+ T cells, impairing their functionality.40
The action of Treg cells may also cause exhaustion of HCV-specific CD8+Tcell response. Treg are T lymphocyte subsets within the CD4+ and CD8+ compartments with strong anti-inflammatory functions. Thus, CD4+ Treg and CD8+ Treg inhibit virus-induced immune activation.41 Their presence is not privative of chronic infection, but when a high-level viremia is persistent, the suppressive activity in vitro is significantly higher than that observed from cells obtained from patients who spontaneously clear the virus.42 Under immune pressure and escaping host defenses, HCV variants emerge. Such viral HLA-II- epitopes variants are able to induce antigen-specific Treg cells to suppress the antiviral T-cell response in an antigen-specific manner, thus attenuating the conventional CD4+ T-cell help required to clear viral infection, and thus promoting its persistence.43
The CD4+ Treg cells are able to directly suppress both the proliferation as well as the IFN-γ secretion of virus-specific CD8+ T cells in vitro. Interestingly, such suppression was not only observed with HCV-specific CD8+ T cells, but also with CD8+ T cells specific for other viruses. However, the in vivo enrichment of the CD4+ Treg cells in the liver might indicate a more specific inhibition on HCV-specific CD8+ T cell by cell-to-cell contact, thus diminishing their immune-mediated damage. This in vivo suppression is inversely correlated with the PD-1 expression level on Treg cells that promotes a lower expansion of intrahepatic Treg cells.44 This local inhibition of Tregs might be viewed as a true contrasuppression, suggesting that a delicate balance between suppressor Treg cells (FoxP3+) and contrasuppressor Tregs (PD-1 + ) might fine-tune this cell population to cut down the harmful responses without switching off those involved in limiting viral spread. In addition, the CD4+Treg cells can indirectly suppress conventional T-cell activity by inhibiting both the DC maturation and the immunostimulatory activity of antigen presenting cell capturing, as well as internalizing and degrading CD80 and CD86 molecules in a CTLA-4 mediated way or, alternatively, by the CD4-homologous molecule lymphocyte activation gene-3 (LAG-3 or CD223), that binds MHC class II molecules.45,46
DC are susceptible to HCV infection and viral proteins (core, NS3, NS4 and NS5) exert a marked, inhibitory effect on their capacity to express HLA and cell surface co-stimulatory molecules, synthesize proinflammatory cytokines such as IL-12 and induce allogeneic T cell proliferation. Compared to DCs purified from peripheral blood, liver DCs produce significantly higher levels of IL-10 and lower proinflammatory cytokine (i.e., IL-1ß, IL-6 and TNF-α) levels. Such HCV-induced impairment on DC function appears to play a key role in the induction and maintenance of HCV-specific Treg cell responses.47–49
Among HCV-infected patients, HCV-specific CD4+ Treg cells are detected in the blood and have the capacity to suppress HCV-specific T cells.50 In liver histological examinations, the CD4+Treg cells are predominantly localized in piecemeal and lobular necroses, in contact with CD8+T cells, infiltrating the hepatic lymphoid aggregates, portal or septal tracts, as well as parenchymatous lobules or nodules. Treg cells are also regulated by the expression of inhibitory signaling pathways. HCV-infected human hepatocytes express high levels of Gal-9 and TGF-β, and upregulate the Tim-3 expression and the regulatory cytokines TGF-ß/IL-10 in conventional CD4+ T cells, converting them into Treg cells.51
As previously mentioned, there are also HCV-specific CD8+ Treg cells that express high levels of IL-10 but its intra-hepatic presence is very scarce.52
During viral infections, IFNs type I (α and β) and III (λ) are produced by infected cells and dendritic cells and IFN type II (−γ) is produced by NK and natural killer T cells as part of the innate immune response, and by antigen-specific T cells (both CD4+Th1 and CD8+ cytotoxic T lymphocytes) representing the adaptive cellular response. Importantly, increased NK degranulation during the acute phase of HCV infection is correlated with the magnitude of the T cell response.53 The NK cells functions are controlled by inhibitory receptors for HLA I, including the killer cell immunoglobulin-like receptors (KIR). HLA presented on NK cells is closely associated with the clearance of HCV and clinical recovery. Patients who have the receptor-ligand combination of being homozygous for KIR2DL3 alleles (which encode KIR2DL3) and HLA-C alleles (which encode its ligand HLA-C1) are more likely to recover from HCV infection and a sustained virological response after combined therapy.54 Interestingly, NK cells in chronic HCV infection have been reported to exhibit impaired IFN-γ secretion by several but not all groups.55–58 Several HCV proteins inhibit host responses when overexpressed in vitro. The HCV core inhibits signal transducers and activators of transcription 1 (STAT1) activation, the NS3/4A protease blocks IFN regulatory factor 3 (IRF3) and impairs the peptidase activities of the proteasome, and the E2 and NS5A proteins inhibit kinase activity of protein-kinase RNA-dependent (PKR). Strikingly, HCV is able to cleave IFN-λ3 by means of its NS34A protease, pointing to the crucial role of such inhibition to favor viral persistence.59
Moreover, NS5A containing apoptotic bodies have been shown to induce monocytes to produce increased levels of IL-10 and decreased amounts of IL-12, leading to a significant downregulation of the activating NKG2D receptor on NK cells via TGF-β.60,61
Paradoxically, IFN-I can also suppress the immune system in ways that promote viral persistence despite a sustained IFN-I signaling, presumably due to ongoing recognition of viral pathogen-associated molecular patterns (PAMPs). In this sense, by using the LCMV chronic infection in mice it has been reported that IFN-I has immunosuppressive capability by triggering PD-1 of activated T cells and increase production of immunosuppressive molecules, including IL-10 and PD-L1.62 Likewise, Lee, et al. showed that 2’-5’ oligoadenylate synthetase (OAS)-like 1 (OASL1) -a member of ISGs- is a novel translation inhibitor of IRF7,the IFN-inducible IFN-I master transcription factor, and negatively regulates robust IFN-I production during acute viral infections.63 The induction of IFN-I negative regulators, such as OASL1 acts early during infection inhibiting both efficient viral control and the induction of functional virus-specific T-cell response, favoring viral persistence and CD8+ T-cell exhaustion. These results indicate that OASL1-mediated suppression of IFN-I production is a critical step for permitting chronic viral infection.64 However, HCV proteins per se did not appear to attenuate IFN-induced HLA class I expression. Instead, in an in vitro cell culture system, the HCV replication is able to suppress IFN-induced HLA class I expression after viral-induced phosphorylation of both PKR and the eukaryotic initiation factor 2α (eIF2α)65 while P56 (ISG56) induction attenuates the induction of IFN-stimulated protein expression in HCV-infected cells. If HCV attenuates IFN-induced HLA class I expression, the role of NK cells would need to be closely monitored. In this connection, NK cells contribute to antiviral innate immune responses by recognizing virus-infected cells that lack HLA class I or overexpress ligands for activating receptors. Although the iminished expression of IFN-induced HLA class I protein in HCV-infected cells can facilitate immune evasion from HCV-specific CD8+ T cells, it might lead to recognition of HCV-infected cells by NK cells. However, HCV is known to evade the NK cell responses in several clever ways, including its direct suppression by the HCV envelope and down-regulation of NKG2D60 avoiding its activation.
PerspectivesThe knowledge on the adaptive immune response to HCV has increased enormously. It is now well established that the functionality of HCV-specific T cells is regulated by a refined network of an expanding repertoire of co-regulatory receptors, which might be harnessed for immunotherapeutic interventions. Since the recent targeting of particular pathways during persistent HCV infections has resulted in variable outcomes, and HCV specific CD8+ T cell exhaustion could be more easily reversed during the acute phase of infection, it would seem worthy to evaluate the effect of targeting multiple co-regulatory pathways at earlier stages than currently explored. Since both liver injury and HCV control are mediated by the host immune system, a better understanding of the mechanisms that explain the dysfunctional T cell response and the viral strategies to evade its effectors is essential to restore such response, thereby possibly limiting the virus infection. Moreover, the viral and host factors that are responsible for the heterogeneity of the strength of the T cell response and the regulatory mechanisms that control HCV-specific immunity will be crucial for the development of effective immune-based therapeutic strategies.
AcknowledgmentWe do apologize to those authors whose findings were not explicitly referenced due to the strict limitation on the number of references that can be cited.
Conflict-of-Interest StatementThe authors declare not conflict of interest.



